Reports: DNI154405-DNI1: Direct Carbon-Carbon Bond Forming Reactions with Hypervalent Iodine Reagents: Redefining Formal SNAr Reactivity with Carbon Nucleophiles
 printer friendly
printer friendlyThe use of diaryliodonium salts as reagents has emerged as a useful strategy for the arylation of diverse nucleophiles, and both metal-catalyzed and metal-free reactions are of significant interest. In metal-free reactions, hypervalent iodine mediates the key aryl-nucleophile bond forming event through a reductive elimination-like pathway. The possibility of a non-metal mediating a reductive elimination-like bond forming event is an intriguing opportunity to expand the scope of nucleophile arylation beyond that of traditional metal-free reactions, namely nucleophilic aromatic substitution (SNAr). This was the premise of our ACS PRF proposal and a central aim of our proposal was the synthesis and use of unsymmetrical aryl(auxiliary)iodonium salts. Specifically, we aimed to determine the extent to which identity of the counter anion and auxiliary influence reactivity and aryl group transfer in metal-free arylation reactions. Despite the vastly superior synthetic utility of unsymmetrical aryl(auxiliary)iodonium salts in chemoselective arylation reactions, the use of symmetrical salts is still common place in the development of new methods. Increased synthetic access of unsymmetrical salts and the study of potentially general auxiliaries would therefore provide much needed information to move forward with unsymmetrical salts.
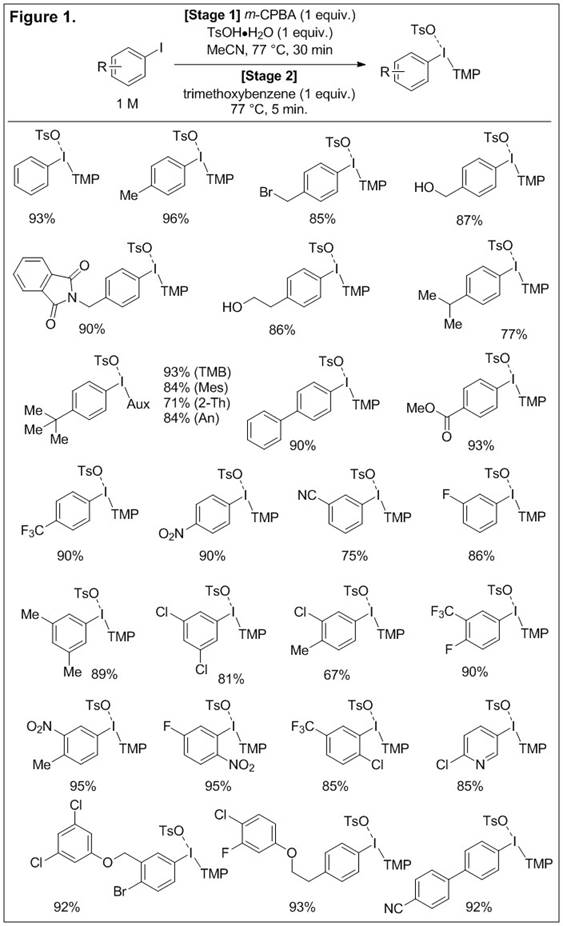
We have focused our efforts, toward this end, on auxiliaries derived from 1,3,5-trimethoxybenzene because there were several reports that indicated its potential as a general auxiliary for arylation of several nucleophiles. However, very few, if any, general methods existed for the synthesis of aryl(2,4,6-trimethoxyphenyl)iodonium salts; none of these reports used commercially available aryl iodides as starting materials. We have described a practical one-pot method that oxidizes the aryl iodide in the first stage and introduces the trimethoxyphenyl (TMP) auxiliary in the second stage. This procedure is straight forward to conduct and is scalable (up to 50 mmol scale). The scope of this transformation is broad and functional group tolerant; greater than 25 examples are provided in Figure 1 and the average isolated yield is greater than 85%. Moreover, based on our use of chemometrics to optimize this reaction we obtained significant insight into the variables that exert the greatest influence on the yield of the reaction. With this information in hand, we were able to replace the toluenesulfonic acid (TsOH•H2O) used in this work with trifluoroacetic acid (TFAH) by reducing the temperature from 77 °C to 50 °C. The switch in acid is not trivial and leads to a direct synthesis of trifluoroacetate salts which are typically obtained by anion exchange of a pre-formed iodonium salt. Finally, Figure 1 contains several compounds with more elaborate aryl moieties that justify the use of unsymmetrical salts as the corresponding symmetrical salts would be much more challenging to prepare and much more wasteful in subsequent arylation chemistry.
We have also described the use of aryl(TMP)iodonium salts in metal-free arylation reactions with C-, N-, O-, and S-nucleophiles (Figure 2). The counter anions used in these reactions are toluenesulfonate (OTs), trifluoromethanesulfonate (OTf) and trifluoroacetate (TFA) which are readily installed by anion exchange or direct synthesis. Moreover, we have found that the aryl(TMP)iodonium trifluoroacetate salts are useful reagents for the arylation of alicyclic amines such as morpholine, pyrrolidine, piperadine, azetidine, and azepane. These heterocycles occur incessantly in drug candidates and the use of hypervalent iodine to mediate the C-N bond forming event has expanded the scope of this reaction beyond SNAr reactions.

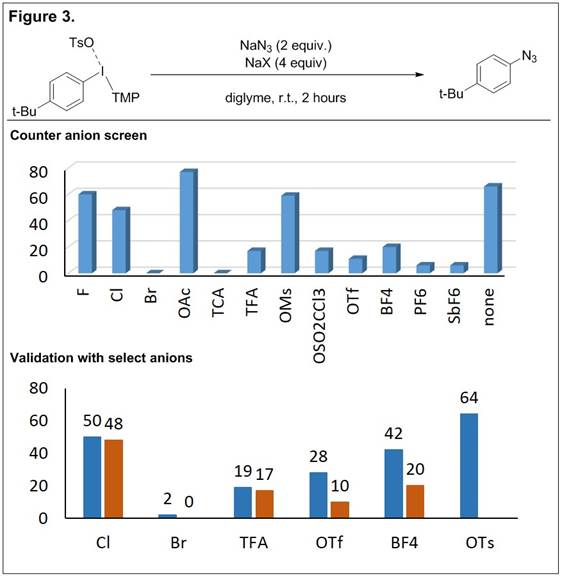
This line of research has also lead to the development of a convenient method to screen counter anions in reaction optimization studies and obviates the need to prepare a range of diaryliodonium salts. In this work the aryl(TMP)iodonium tosylate serves as a key starting material and the counter anions of interest are doped into reactions as the sodium salts (Figure 3). An in situ anion exchange of tosylate for the doped counter anion provides a facile method to screen a diverse range of counter anions. The doping protocol was verified with select counter anions in which reactions with the preformed salt (Figure 3, “validation”, blue) provided similar yield to the doping protocol (Figure 3, “validation”, red). The method was developed for an arylation of azide but we have also demonstrated its applicability with boron, carbon, and oxygen nucleophiles.
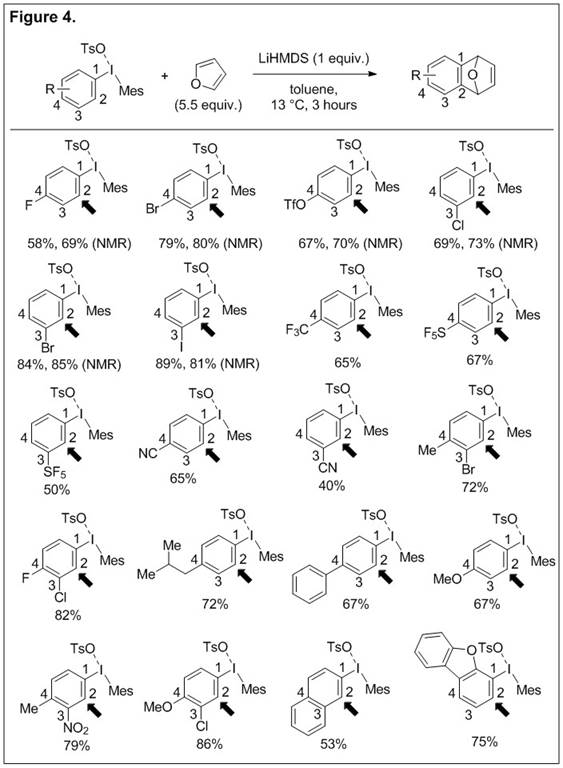
The direct ipso-substitution of the iodonium moiety in diaryliodonium salts is the dominant pathway in reactions with nucleophiles. During our study of these reactions we have also uncovered another pathway that leads to aryne intermediates and this reactivity has been developed into the first high-yielding route to arynes from diaryliodonium salts via C-H deprotonation. Unsymmetrical aryl(mesityl)iodonium salts are novel aryne precursors because they are readily available from simple arenes, aryl iodides, or arylboronic acids in one-pot reactions and they may be isolated as analytically pure solids by trituration from the reaction mixture with ether. We have demonstrated that these arynes may be trapped with furan in a cycloaddition reaction (Figure 4). The C-H deprotonation event is regioselective (Figure 4, indicated arrow) and the ejection of the iodonium leaving group is highly chemoselective in the presence of other halides or pseudohalides. The reaction tolerates electron donating and withdrawing substituents on the aromatic ring as well as ortho-, meta-, and para-substitution.
This ACS PRF award has, in part, provided the funds necessary to initiate my research program in organic synthesis methodology. Specifically, stipend support for students was a critical component to establishing the feasibility of the chemistry and developing the reactions described above. Over the total funding period three graduate students and one undergraduate received stipend support to contribute to various aspects of the projects. The training of postdocs and students in organic synthesis and reaction methodology totaled one postdoc, four graduate students, and 2 undergraduates.










