Reports: UR453688-UR4: Transition State Analogues as Mechanistic Probes for Asymmetric Desymmetrization by Cinchona Alkaloid-Based Catalysts
 printer friendly
printer friendlyIntroduction
The goal of this project is to model the intermolecular forces governing enantioselectivity in the asymmetric desymmetrization (ASD) of cyclic anhydrides (Fig. 1).[1] This reaction introduces chirality into inexpensive achiral feedstocks, and has been applied in the total synthesis of biologically active compounds.1,[2] The model we develop can then be used to design or screen potential catalysts, in order to achieve high selectivity with a range of substrates.

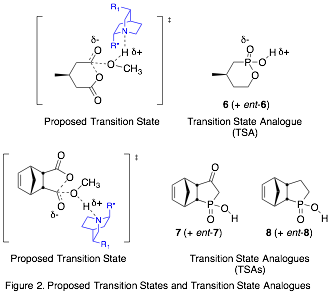
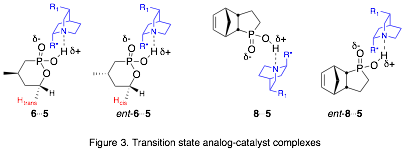
Our approach is to prepare transition state analogues (TSAs) for the organocatalytic ASD reaction and study the interactions between the TSAs and the catalysts, which are cinchona alkaloid derivatives.[3],[4] We are using tetrahedral phosphonic or phosphinic acids as TSAs to mimic the transition state and hydrogen bond to the catalyst (Fig. 2). To evaluate our model, we will study the interactions of the TSA enantiomers with catalyst. Furthermore, we will measure the relative binding affinities of the enantiomers with catalysts and correlate those with the enantiomeric excess (ee) of the ASD reaction (Fig. 3).
Goals
1. Synthesize TSAs and resolve into pure enantiomers.
2. Evaluate catalyst-TSA interactions by NMR spectroscopy and X-ray crystallography.
3. Measure relative binding of two enantiomers of TSA to catalysts and compare with ee values.
1. Synthesis
Both R and S enantiomers of TSAs 6 and 8 have been prepared in 90% ee or greater, as described in our 2015 report.
The 2015 report recounted the synthesis of the methyl ester of 7, which has been more challenging to deprotect than the methyl ester of 8. Electrophilic reagents such as TMSBr have led to decomposition, and basic reagents such as hydroxide result in enolization instead of deprotection. However, lithium iodide in acetonitrile enables >90% conversion to the lithium salt of the desired product. Ion exchange of the crude reaction product results in product decomposition; therefore, we are currently attempting other means of purification to complete the synthesis of racemic 7. This will be resolved into pure enantiomers in the same way as TSA 8.
Future plans:
Assign the configuration of enantiomers of TSAs 7 and 8 by X-ray crystallography.
2. Catalyst-TSA interactions
We are using NOESY NMR to study catalyst-TSA interactions. We have observed intermolecular NOEs between a proton of TSA 6 and ent-6 (colored red in Fig. 3) and some aromatic and quinuclidine protons of 5a (colored red in Fig. 1). Importantly, these NOEs are observed between the proton trans to the methyl group for 6 and cis to the methyl group for ent-6, indicating that the two enantiomers approach the catalyst from opposite sides of the six-membered ring. This suggests that hydrogen bonding via the phosphonic acid group controls the orientation with which the TSA approaches the catalyst and is consistent with our overall hypothesis. We are currently performing similar NOE studies with catalyst 5b and we are crystallizing each TSA enantiomer with catalysts 5a and 5b, in order to obtain X-ray structures of these interactions.
Future plans:
Perform comparable NMR and crystallographic studies of 8 and ent-8 with catalysts 5a and 5b, as well as develop theoretical models of the catalyst-TSA interactions using computational methods.
3. Relative binding strength
Another goal of this project is to correlate the relative binding strength between the catalyst and each TSA enantiomer with the ee of the ASD reaction. We have used DOSY NMR to obtain diffusion coefficients (D) of the catalyst-TSA complexes at different concentrations of TSA.[5],[6] What we have found is that the measurement error for D values is greater than the difference in D between samples. We are currently measuring D values of samples that combine catalysts 5a or 5b with racemic samples of 6 or 8. In order to resolve the NMR signals of the two enantiomers of 6, we use 1:1 molar ratios of 6:5b. Under these conditions, the D values for all species in solution are identical. In contrast, we do see a difference in D value for the two enantiomers of TSA 8 and catalyst 5a, under conditions where the enantiomer NMR signals are resolved. At this time, we have not identified the absolute configuration of the species that binds more tightly.
Future plans:
Vary concentrations and catalyst:TSA ratios to resolve enantiomer resonances in NMR for measuring D values in the presence of catalysts.
Impact on Students
A total of five students have done summer research supported by the ACS-PRF award (one for two consecutive summers). Their experience includes enantioselective synthesis and chiral resolution, and multi-nuclear and two-dimensional NMR. All students give a formal talk to the chemistry department, a poster presentation at Carleton, write a formal report, and continue research during the academic year. Three students have presented their results at a national ACS meeting (poster). One of the two graduates is enrolled in a PhD program at Northwestern University, one is applying to graduate programs and one is applying to medical school for expected enrollment in 2017.
Impact on PI
The PRF award was excellent leverage for my successful NSF-MRI proposal (with two co-PIs) for a new NMR spectrometer. The project has also provided me with excellent experience in DOSY NMR.
[1]. Borissov, A.; Davies, T. Q.; Ellis, S. R.; Fleming, T. A.; Tichardson, M. S. W.; Dixon, D. J. Chem. Soc. Rev. 2016, Ahead of print.
[2]. Diaz de Villegas, M. D.; Galvez, J. A.; Etayo, P.; Badorrey, R.; Lopez-Ram-de-Viu, P. Chem. Soc. Rev. 2011, 40, 5564-5587.
[3]. Oh, S. H.; Rho, H. S.; Lee, J. W.; Lee, J. E.; Youk, S. H.; Chin, J.; Song, C. E. Angew. Chem. Int. Ed. 2008, 47, 7872-7875.
[4]. Chen, Y.; Tian, S.-K.; Deng, L. J. Am. Chem. Soc. 2000, 122, 9542-9545.
[5]. Fielding, L. Tetrahedron 2000, 56, 6151-6170.
[6]. Dethlefs, C.; Eckelmann, J.; Kobarg, H.; Weyrich, T.; Brammer, S.; NŠther, C.; LŸning, U. Eur. J. Org. Chem. 2011, 2066-2074.










