Reports: DNI154007-DNI1: Rhodium-Catalyzed Amide Synthesis
 printer friendly
printer friendlyThe PRF grant was used to support two different projects during 2015-2016, the originally funded development of a titanium- or zirconium-oxo mediated alkyne aldehyde cross coupling reaction for the synthesis of α,β-unsaturated carbonyl compounds and for the Rh-catalyzed oxidative amidation reaction.
Ti/Zr=O Mediated Alkyne-Aldehyde Cross Coupling
α,β-Unsaturated aldehydes and ketones are both prominent functionalities in and synthetic precursors for pharmaceuticals, natural products, and organic materials. Olefination reactions are powerful methodologies for the synthesis of enones; unfortunately, these reactions often require several synthetic steps to access the requisite starting materials and generate stoichiometric waste. The goal of the proposed research is to develop a new transition metal-catalyzed cross-coupling reaction for the synthesis of α,β-unsaturated carbonyls from readily available building blocks without the generation of any stoichiometric byproducts in a redox-neutral, atom-economical process. This should be possible by employing a cycle which uses a Ti/Zr=O catalyst to couple a carbonyl with an alkyne (Figure 9); through, (i) [2+2]-cycloaddition between the [M]=O and an alkyne, (ii) insertion of a carbonyl into the [M]=C bond of the oxametallacyclobutene, and, finally, (iii) [4+2]-retrocycloaddition, releasing the α,β-unsaturated carbonyl product and regenerating the [M]=O catalyst. As the starting materials are already at the requisite oxidation states, the reaction will not require either a stoichiometric oxidant or reductant.
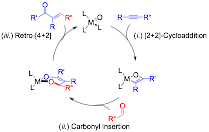
Figure 1 Proposed alkyne-aldehyde cross coupling.
During the prior years funding we demonstrated that Zr was able to mediate the desired reaction stoichiometrically but that the transformation required high temperatures (140 °C) and was theromodynamically disfavored, requiring an external reagent, chalcone, to trap the Zr=O complex as the retro-[4+2] was reversible. Our efforts then focused on the development of an analogous reaction mediated by Ti. We were able to demonstrate that the [2+2] reaction was facile and regioselective and that the retro-[4+2] occurred at room temperature in the presence of a Lewis base (4-(dimethylamino)-pyridine) (Table 1) indicating that Ti would be the superior catalyst for the transformation.
Table 1 [2+2]-Cycloaddition Scope
![]()
Entry |
Cpx |
R' (product) |
Yielda |
1 |
Cp* |
Ph= |
95% |
2 |
Cp* |
p-MeO-C_6H4= _ |
92% |
3 |
Cp* |
3,5-(CF3)2-C_6H3= |
88% |
4 |
Cp* |
p-F-C6H4= |
70% |
5 |
Cp* |
p-NO2-C6H4= |
0%b |
6 |
Cp' |
3,5-(CF3)2-C_6H3= |
94% |
Unfortunately, the carbonyl insertion step proved to be the downfall of the desired reaction, as it was not observed with a variety of ligands, carbonyl compounds, and alkynes (Figure 2). Rather, other deleterious side reactions occurred. We did take the opportunity to develop new and improved synthesis of Ti-oxo complexes, investigate the reactivity of their reactivity, and the mechanism of the retro-[4+2] cycloaddition (Figure 3). The findings of these studies are disclosed in our recent Organometallics full paper.
![]()

Figure 2 Proposed decomposition pathway of the Ti-oxo
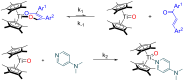
Figure 3 Mechanism of the retro-[4+2] cycloaddition
Rhodium-Catalyzed Oxidative Amidation
New approaches for the synthesis of amides were identified as a key research area for Green Chemistry in 2007. We seek to develop a chemoselective Rh-catalyzed amide bond forming reaction that generates this important functionality through a transfer hydrogenation reaction between amines and aldehydes, in situ generated aldehydes, or aldehyde equivalents. Eventually, we also seek to expand this method to the synthesis of esters, thioesters, and ketones through the use of oxygen, sulfur, and carbon nucleophiles, respectively (Scheme 1). Finally, in addition to the formation of the desired carbonyl functionality, chiral catalysts will be developed which allow for the formation of proximal stereocenters.
Over the past year, our efforts on the development of the Rh-catalyzed oxidative functionalization have successfully developed conditions for the oxidative esterification reaction (Figure 4). Further, as a chiral catalyst is used, the β-stereocenter is established with high enantioselectivity (> 90% ee). It is important to note that the alcohol is only required in a moderate excess, rather than in solvent quantities common to other oxidative esterification reactions. The methodology works with an array of alcohols, allylic amines, and allylic alcohols. Currently, the scope is being flushed out, products isolated with a paper submission in the near future.

Figure 4 Rh-catalyzed esterification
We have also developed an oxidative amidation reaction of sterically hindered aldehydes and alcohols. These substrates are challenging to other oxidative amidation catalysts, as the corresponding amide is formed in low yield, if at all. Additionally, anilines have proven to be challenging nucleophiles. We have found that Rh is an excellent catalyst for the synthesis of amides adjacent to quaternary stereocenters with both secondary amine, primary amine, and aniline nucleophiles (Figure 5). In addition to exploring the scope of the transformation we have conducted preliminary mechanistic investigations. This work is currently under review in ACS Catalysis.
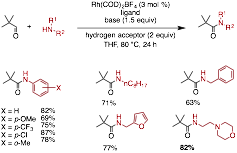
Figure 5 Examples of the oxidative amidation of sterically hindered aldehydes
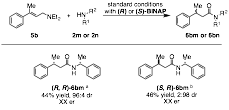
Finally, we have successfully developed the asymmetric oxidative amidation of allyl amines for the synthesis of β-chiral amides. We found that prochiral allylic alcohols underwent the oxidative amidation reaction to afford the desired amide in low enantioselectivity. However, allyl amines are known to undergo a related isomerization with excellent levels of enantioselectivity; if diethylallyl amine is used, the enamine undergoes exchange with external nucleophiles allowing a common allyl amine to generate an array of enantiopure (>90% ee) amide products (Figure .

Figure SEQ Figure \* ARABIC 6 Rhodium catalyzed oxidative amidation of diethyl allyl amine
Under our optimized reaction conditions we were able to demonstrate that the methodology can be applied to the synthesis of both secondary and tertiary enantiomerically enriched β-chiral amides (Figure 7). Further, when the reaction is performed on chiral allyl amines, the diastereoselectivity is catalyst controlled, rather than substrate controlled, to afford either diastereomer in excellent stereoselectivity and good to excellent yields (Table 2). Finally, when chiral amines are employed no racemization is observed (Figure 8). 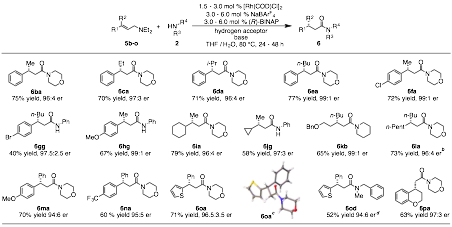
Figure 7 Scope of the asymmetric oxidative amidation.
Table 2 Catalyst controlled diastereoselective oxidative amidation
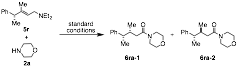
|
entry |
Ligand |
Yield a |
dr (6ra-1/6ra-2) b |
|
1 |
(R)-BINAP |
46% |
96.5 : 3.5 |
|
2 |
(±)-BINAP |
67% |
14 : 86 |
|
3 |
(S)-BINAP |
73% |
2:98 |
|
|
|
|
|
Figure 8 Catalyst controlled diastereoselective amidation