Reports: DNI155070-DNI1: Light-Driven Dehydrogenative Carbon-Carbon Coupling Reactions
 printer friendly
printer friendlyPRF Grant Narrative Progress Report
Photoredox catalysis has recently emerged as a powerful strategy for introducing kinetically facile single electron pathways into a broad range of useful organic transformations. The most common photosensitizers used in these reactions are molecular d6 octahedral complexes of Ru and Ir, which can act as one-electron donors or acceptors in their excited states. Because these photosensitizers function by outersphere electron transfer, the scope of transformations is restricted by the need to balance the redox potentials of each starting material, intermediate, and product in order to achieve a viable catalytic process. In contrast to their molecular counterparts, heterogeneous photosensitizers, commonly used in energy catalysis, have rarely been studied in organic reactions. In principle, the ability to exploit specific interactions between organic substrates and catalyst surface active sites might provide orthogonal tools to build selectivity into photoredox reactions.
TiO2 is a large-band gap semiconducting material. Upon excitation with UV light, TiO2 is capable of delivering hole equivalents near 3.0 V vs. NHE, a potential that is sufficiently oxidizing to generate hydroxyl radicals from water. Because of its potent oxidizing power, TiO2 has been investigated in applications such as water and air purification, where the complete degradation of all organic compounds to CO2 is desired. Recently, the highly oxophilic nature of TiO2 has been exploited to bind alcohol substrates and enable their selective dehydrogenation to aldehydes under illumination with visible light. For example, Tanaka reported the conversion of 2-propanol to acetone using a series of metal/TiO2 catalysts. In this progress report, we describe our efforts over the past year to apply this basic alcohol dehydrogenation reactivity to organic transformations of imines.
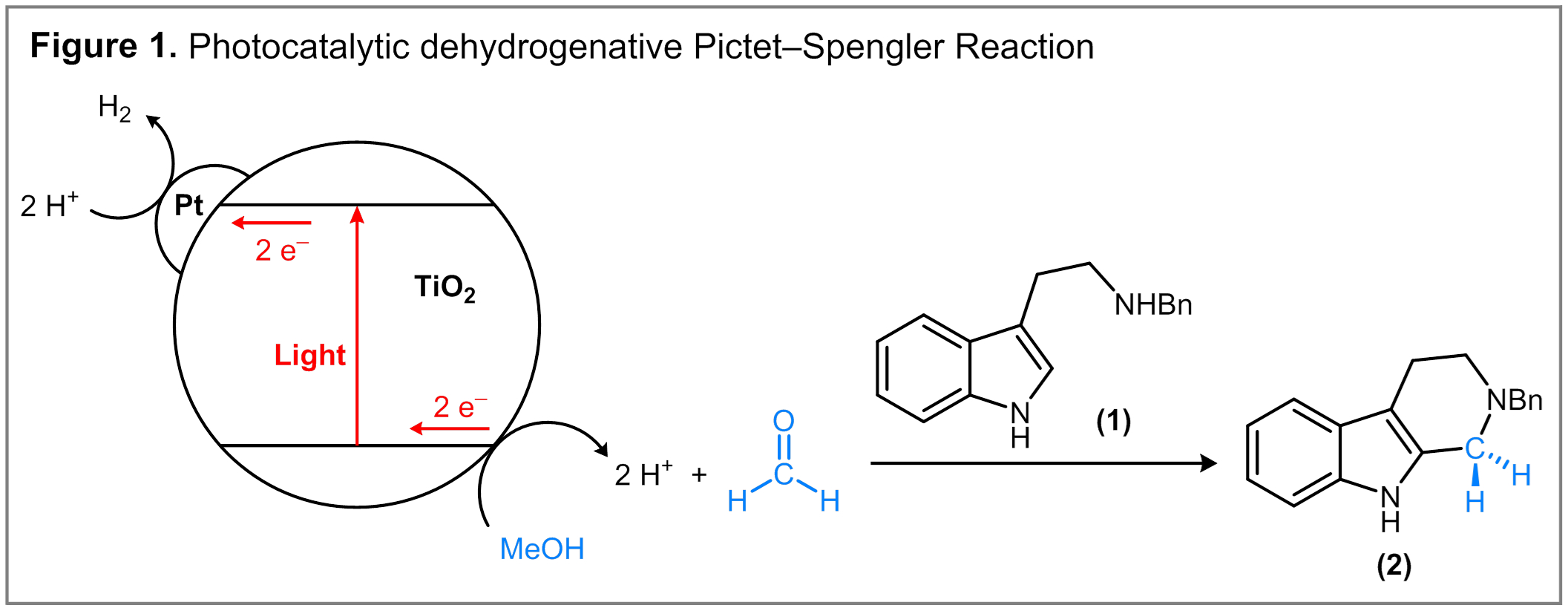
We initiated our studies by examining the Pictet-Spengler cyclization of N-benzyltryptamine (1) as a model reaction. A proposed mechanism is illustrated in Figure 1. Visible light irradiation of Pt/TiO2 particles result in electron–hole separation. The electrons are scavenged by the Pt co-catalyst and used to convert protons to gaseous H2. The hole equivalents are transferred to surface-bound alcohol molecules to produce aldehydes. Condensation of the aldehyde equivalents with tryptamine yields an iminium ion intermediate, which subsequently undergoes cyclization to form a tetrahydro-b-carboline product.
Accordingly, a 0.5 wt% Pt/TiO2 catalyst effects the conversion of 1 in MeOH to form 2 in 90% isolated yield upon irradiation with a 100 W Hg lamp under an N2 atmosphere. The addition of AcOH (2.0 equiv) was found to provide optimal yields of 2 and minimizes the formation of N-methyltryptamine, a substantial side product in the reaction. The formation of H2 as the terminal reduction product was confirmed by mass spectrometry analysis of the headspace gas formed in the reaction. Additionally, the use of isotopically labeled CD3OD yielded 2-d2 and gaseous D2. Under aerobic oxidation conditions, the same Pt/TiO2 catalyst is capable of generating 2 but does so in a relatively low yield of 52%.
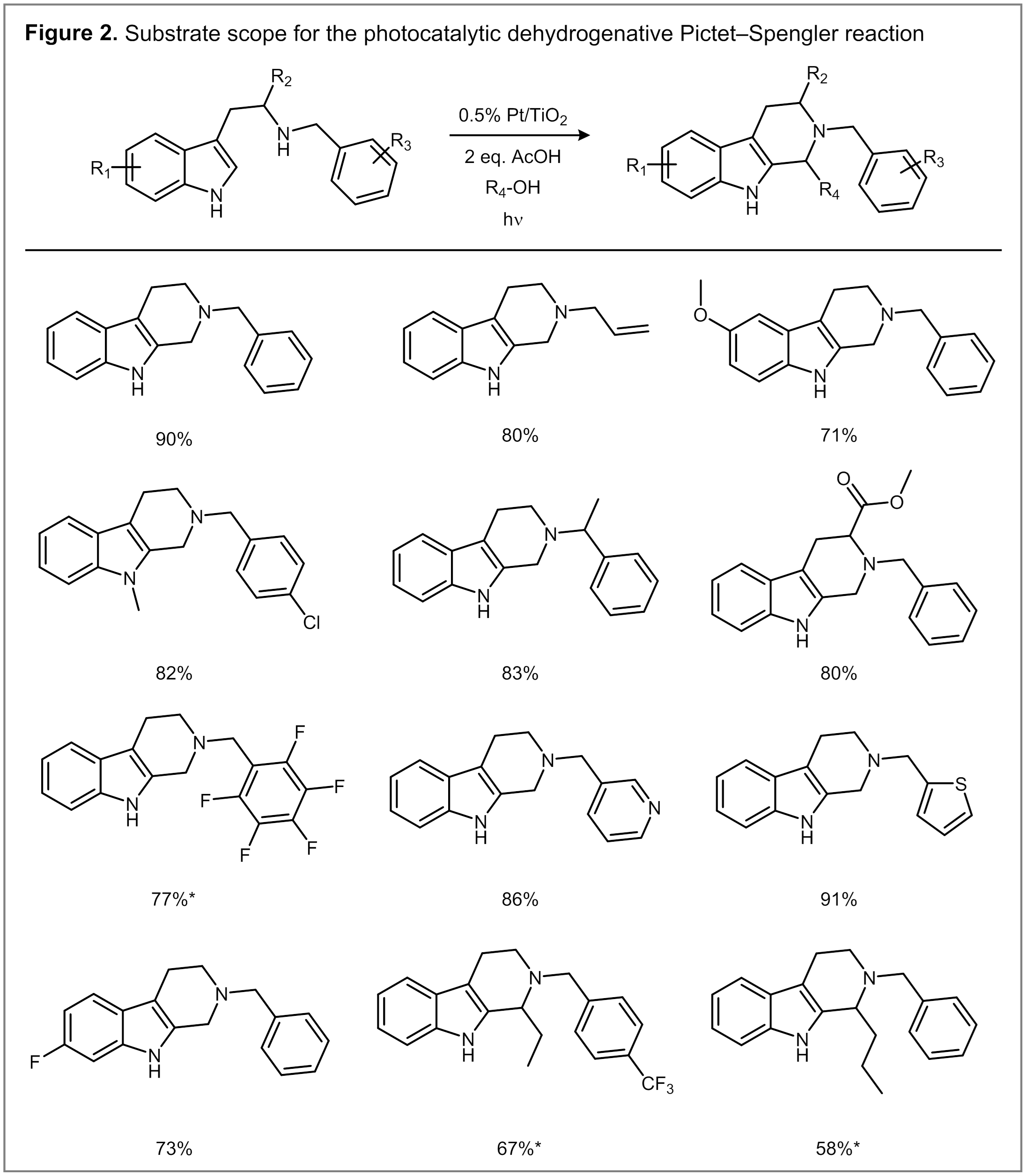
The reaction exhibits a broad scope with respect to substitution on the tryptamine substrate (Figure 2). A range of electron-withdrawing and donating substituents are tolerated on the N-benzyl group. Notably, p-methoxybenzyl can be used despite its susceptibility to C–N bond cleavage under oxidizing conditions. Various substrates containing substituted indole rings also react with similar efficiency. The reaction tolerates substitution on the ethylene bridge in the form of tryptophan methyl ester. Finally, the use of higher primary alcohols, such as ethanol or 1-butanol, provides access to Pictet-Spengler products bearing substitution adjacent to the indole ring.
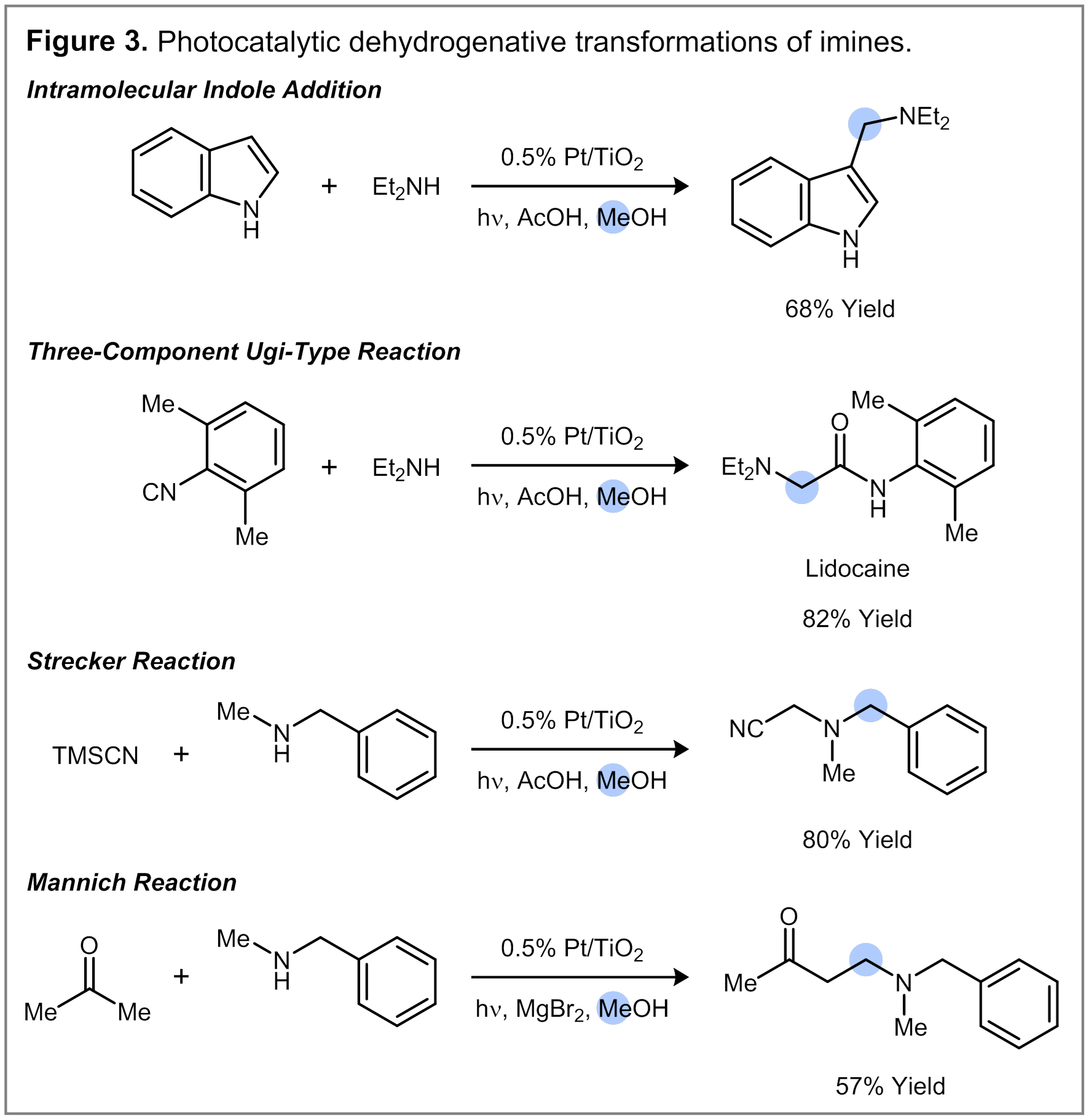
The photocatalytic alcohol dehydrogenation can be exploited in other imine transformations relevant to organic synthesis (Figure 3). For example, the intramolecular reaction between indole and Et2NH in MeOH generates the corresponding 3-aminomethylindole product in 68% yield. Isonitriles and amines undergo an Ugi-type three-component reaction to generate -aminoamide products. For example, the Pt/TiO2-mediated reaction between diethylamine and 2,6-dimethylphenylisonitrile in MeOH solvent provides Lidocaine in 82% isolated yield. Strecker and Mannich reactions can also be carried out to form aminonitrile and b-aminoketone products respectively.
Ongoing work is aimed at fully developing the scope of these dehydrogenative coupling reactions and extending these concepts to other classes of heterogeneous catalysts and transformations.
Impact. During the past year, funding from the ACS PRF grant was used to support one graduate student during the summer and one graduate student during the spring 2016 semester. Students were able to use this opportunity to carry out much of the reaction optimization, substrate scope studies, mechanistic work, and materials characterization studies described in this report. Additionally, a student travelled to the 2016 Central Regional Meeting of the ACS to present a seminar. This work is currently being written up for publication.










