Reports: DNI154831-DNI1: Divergent Access to Hydrosilylation and Dehydrogenative Silylation Reactions Exploiting Grubbs-Type Ruthenium Benzylidene Complexes
 printer friendly
printer friendlyNon-metathetical applications of Grubbs-type ruthenium complexes have been extensively developed as new synthetic strategies. For decades, many powerful metal-catalyzed hydrosilylation, which is one of the most important homogeneous catalytic processes, have been developed for preparation of materials with significant functions. However, ruthenium alkylidene complex-catalyzed alkene hydrosilylation to provide silicon-containing heterocycles has not been reported. During the previous funding period, our efforts focused on the development of the efficient synthetic strategy to access valuable organosilanes exploiting Grubbs-type ruthenium complex-catalyzed intramolecular alkene hydrosilylation of alkenylsilyl ethers. We initiated this project by questioning whether Grubbs-type ruthenium complexes can selectively promote intramolecular alkene hydrosilylation over alkene metathesis. We recognized several potential challenges of the ruthenium alkylidene-catalyzed alkene hydrosilylation, some of which include 1) lower reactivity of ruthenium catalysts toward hydrosilylation compared with other late transition metal catalysts, 2) the potential homo-dimerization metathesis, and 3) lack of detailed mechanistic studies. To the end, we demonstrated non-metathetical use of such ruthenium complexes for alkene hydrosilylation via preferential Si–H bond activation over alkene activation. Importantly, this study has led to a better understanding of fundamental mechanistic aspects of non-metathetical function of Grubbs-type ruthenium catalysts for alkene hydrosilylation. We proposed direct s-bond metathesis between Si–H and Ru–Cl via a four-centered transition state within a ruthenium coordination sphere to afford a putative ruthenium silane complex in the initial stage of hydrosilylation, which substantially differs from the previously proposed Chauvin-type mechanism, that is, the addition of R3Si–H across the p-bond of a Ru-benzylidene complex (Figure 1). The ruthenium alkylidene-catalyzed alkene hydrosilylation also proceeds the modified Chalk-Harrod mechanism (i.e., silylruthenation) rather than the Chalk-Harrod (i.e., hydroruthenation) pathway. The lesson can be potentially expanded toward for other ruthenium alkylidene-medicated transformations such as dehydrogenative condensation between alcohols and silanes, direct C–H arylation, and hydrogenation of unsaturated C–C double bonds. In addition, we investigated the scope of the process and regio- and stereoselectivity of the hydrosilylative (5-endo-trig and 6-exo-trig) cyclization (12 examples). We have completed this Aim and published this work in ACS Catalysis 2015.

Figure 1. Intramolecular Alkene Hydrosilylation Exploiting Grubbs-Type Ruthenium Alkylidene Complexes
II. Dehydrogenative Silylation and Hydrosilylation of Vinyl Arenes Catalyzed by Ruthenium Alkylidenes
A second aim of emphasis during this time period, we have been developing selective dehydrogenative silylation and hydrosilylation of vinyl arenes exploiting ruthenium alkylidene catalysts to access vinylsilanes and alkylsilanes. Both relatively stable and virtually non-toxic vinylsilanes and alkylsilanes have been important synthetic building blocks for pharmaceutical targets and materials. Although such organosilanes have been extensively utilized as valuable synthetic intermediates through subsequent conversions of silicon moieties, regio- and stereoselective dehydrogenative silylation of alkenes to afford vinylsilanes are challenging, due to either competing processes involving hydrosilylation to provide alkylsilanes or b-hydride elimination to give allylsilanes. Nevertheless, direct silylation of more accessible alkenes than alkynes to afford vinylsilanes is more desirable. The previous approaches for dehydrogenative silylation of alkenes generally utilized an excess sacrificial hydrogen acceptor (SHA) and either excess alkenes or silanes. Furthermore, such methods require air- and moisture sensitive catalysts, or more reactive alkylsilanes instead more versatile alkoxysilanes. Thus far, Grubbs-type ruthenium complex-catalyzed dehydrogenative silyl coupling of alkenes and alkoxysilanes to provide vinylsilanes is unknown. To the end, we achieved selective dehydrogenative silylation and hydrosilylation of a virtually equimolar ratio of vinyl arenes and alkoxysilanes exploiting ruthenium alkylidene catalysts to access only (E)-vinylsilanes and alkylsilanes (Figure 2). Primarily, regio- and stereoselective access to either dehydrogenative silylation or hydrosilylation was realized simply by varying ligand structure of ruthenium alkylidene catalysts, specifically L- and X-type ligands at ruthenium. For example, we demonstrated the dehydrogenative silylation exploiting the readily accessible Grubbs first-generation catalyst (X-type ligand = Cl and L-type ligand = PCy3), with moderately ROMP-active cycloalkenes (e.g., cis,cis-1,5-cyclooctadiene) as a sacrificial hydrogen acceptor. We explored the scope of ruthenium alkylidene-catalyzed dehydrogenative silylation of styrenes with Ru-1, which exhibited relatively broad functional group tolerance and high regio- and stereoselectivity to afford (E)-vinylsilanes (27 exmaples). We then investigated the scope of ruthenium alkylidene-catalyzed hydrosilylation of styrenes and HSiMe(OTMS)2 with Ru-7, best known for Z-selective olefin metathesis catalysts, containing an NHC L-type and bidentate nitrate X-type ligands as well as chelating adamantyl ligand. This process provided moderate to good yields of alkylsilanes (14 examples) and exhibited good functional group tolerance and good product selectivity. Our preliminary mechanistic studies on dehydrogenative silylation showed that the turnover-determining step is the Si–H cleavage by ruthenium alkylidene catalyst to produce the putative ruthenium silyl complex and HCl, which is consistent with observation of ruthenium alkylidene as the resting state. This work was submitted to Organic Letters and the paper has been just accepted. We are currently investigating the origin of such ligand-controlled selectivity regarding dehydrogenative silylation and hydrosilylation as well as their detailed mechanism.
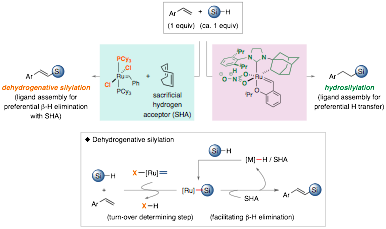
Figure 2. Ruthenium Alkylidene-Catalyzed Dehydrogenative Silylation and Hydrosilylation of Vinyl Arenes
III. Synthetic Applications of Silyl Acetals and Catalysis
A final area of study was synthetic applications of silyl acetals. We first developed a single-pot, catalytic reductive C–H silanolization of aromatic esters with silyl acetal directing groups. Then, we established arene ortho-C–H silylation of phenols with traceless, versatile, acetal directing groups and demonstrated synthetic applications of the resulting dioxasilines. Additionally, silyl acetals were utilized to a Lewis base-promoted, reductive Horner-Wadsworth-Emmons olefination, which addressed the long-standing issue of the reaction with challenging substrates including aryl, alkenyl, and alkynyl esters. We have recently reported the results to ChemComm 2015, JOC 2015, OL 2015, JACS 2016.
In summary, the ACS PRF supports to our research program has provided research training for students and allows us to develop several new research programs. Through this research opportunity students learned and equipped necessary synthetic skills. During this funding period we reported 6 publications and we expect two additions publications, which will have resulted from our future efforts.










