Reports: ND153765-ND1: Gold-Catalyzed Photoredox Transformations
 printer friendly
printer friendlyOverview: To mimic Nature's photosynthesis, chemists have developed a variety of photoredox complexes. Among theses, polypyridine Ru(II) and Ir(III) complexes (1-3) possesses high-energy, long-lived and highly emissive excited states to be useful in organic synthesis (Figure 1). While these complexes generate a variety of synthetically useful carbon-centered radical intermediates, however they suffer from low reduction potentials inherent to each complex (-1.31 V to -1.73 V versus SCE), limiting radical intermediate scope to those derived from activated C-X bonds. With these limitations in mind, it is important to develop new photoredox catalysts that offer the ability to reduce unactivated carbon−halogen bonds with higher reduction potentials, thus giving access to a wide range of organic free radicals from readily available starting materials. Taking advantage of exciplex formation, dimeric Au(I) complexes are capable of generating carbon-centered radicals from unactivated bromoalkanes and arenes (> -2.4 V vs. SCE) that have redox potentials far above that of the photocatalyst.
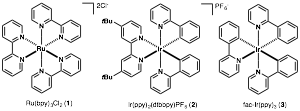
Figure 1
Progress Report: In the outlook and future plan of the previous progress report (2014-2015), we asked this question: 'Does the oxidative or reductive quenching cycle operate in an inner or out-of-sphere mechanism?' To answer this question, we have synthesized various gold photocatalyst complexes (I-V) and performed a series of quenching experiments (in collaboration with Prof. Tito Scaiano, uOttawa). These experiments have shed more light on the mechanism involving the oxidative and reductive quenching cycles of excited [Au2(dppm)2]Cl2 (Figures 2A and B). Upon excitation of the dimeric Au(I) species, exciplex formation with a bromoalkane triggers an oxidative quenching of the photocatalyst (Figure 2A); a SET event leading to an intermediate [AuI-AuII]3+ complex and carbon centered radical genesis. The incumbent radical can undergo coupling reactions with radical acceptors, where the intermediate formed can reduce the [AuI-AuII]3+ complex, thus regenerating the ground state photocatalyst and providing an alkyl functionalized product in a redox-neutral manner. In the reductive quenching cycle (Figure 2B), the excited state photocatalyst first triggers SET between an trialkylamine (e.g. N,N-diisopropylamine (DIPEA)) leading to an intermediate [AuI-Au0]+ complex and a radical cation of the trialkylamine. The intermediate [AuI-Au0]+ complex is a powerful reducing agent that can activate bromoalkanes/arenes through an SET event, regenerating the ground state photocatalyst and generating carbon centered radicals. These radicals can then undergo radical reactions and provide alkyl/aryl functionalized products. In general, the oxidative quenching pathway is the major pathway only when trialkylamines are absent from the reaction medium when using [Au2(dppm)2]Cl2 (complex I). It has been shown that trialkylamines quench the excited state catalyst approximately 10X faster than bromoalkanes (2.7 x 107 M-1s-1 for DIPEA vs 2.9 x 106 M-1s-1 for n-BuBr) (Figure 3C). However, by modification of the phosphine ligands, the trialkylamine and bromoalkane quenching rates for dimeric Au(I) complexes may be modulated. We found that relatively donating ligand bis(diphenylphosphino)methane (dmpm, complex II) to dppm leads to a switch in selectivity of the excited state quenching, ~40 fold faster quenching of for n-BuBr than DIPEA. When using an electron withdrawn trifluromethylated dppm ligand, the excited state of complex III is quenched by DIPEA is nearly 4 orders of magnitude faster than n-BuBr. Hence, more electron-donating ligands lead to more reducing complexes (reacts with bromoalkane first). This is exemplified by carbene ligated gold complex V and is closely reflected by the redox potentials reported for each complex.
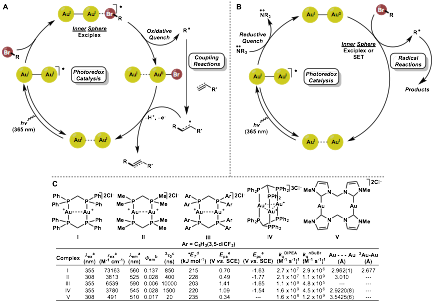
Figure 2
Following this study, the prospects of activating sp2 C-Br bonds for intramolecular addition reactions were evaluated. β-Bromoenones 1-3 were shown to be interesting sources of a rare vinyl radical intermediate, where efficient cyclization onto pendent arenes provided butenolide polycyclic cores 4-6 in good yields (Figure 3A). As shown in Figure 3B, a range of 3-bromocyclohexenones 7-9 were also evaluated. These interesting precursors gave rise to an efficient route to functionalized polycyclic naphthol cores 10-12, up to 69% yield. The utility of this transformation was further demonstrated by achieving a concise formal synthesis of (±)-triptolide 16. The cascade cyclization of 13 gave 14 which upon treatment with sulphuric acid provided the tetracycle 15 in 64% yield. The latter was converted in few steps to the natural diterpene (Figure 3C).
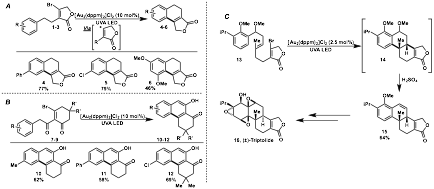
Figure 3
We have revisited our initial report of hydrodebromination of bromoalkanes and haloarenes using UVA LEDs instead of sunlight with the [Au2(dppm)2]Cl2 catalyst. Surprisingly, irradiation of (17-21) with UVA LED led to dimerized by-products (28-32) rather than the reduced products observed using sunlight. Optimization of this protocol with deuterated solvents and TEA led to the dimerization of a few primary and secondary bromoalkanes including substrate 20 prone to 5-exo-trig cyclizations before dimerization (Figure 4A). Furthermore, this protocol was applied to iodoarenes such as 22-26, leading to biaryl products 33-38. A variety of electron deficient and neutral iodoarenes were coupled in good yield including iodo-functionalized phenylalanine and estrone derivatives 26 and 27 (Figure 4B). Mechanistic studies revealed that free radicals may be important intermediates in this reaction but due to low concentrations, radical-radical recombination is unlikely (Figure 4C). For this reason, it was proposed that an gold atom of the photocatalyst may be acting as an acceptor of alkyl radicals, where upon high enough concentration, may undergo an Au(I)/Au(III) reductive elimination pathway which is a scarcely found pathway.
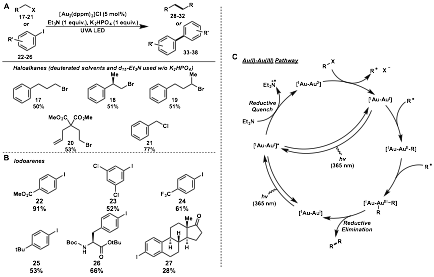
Figure 4
Conclusion
This grant has allowed our group to explore and innovate toward the development of novel light-enabled organic transformations. Herein, we have described dimeric Au(I) complexes as photocatalysts used in the blossoming field of photoredox catalysis. These findings have paved the way to important contributions in photoredox and recently in the rarely accessible Au(I)/Au(III) catalytic cycle. Since our original publication in 2013, other groups have used our catalyst and approach to develop new transformations. These studies have also generated new questions of the structure and reactivity of these Au complexes and their mechanistic pathways. With this in mind, one can envision immense possibilities in the development of new transformations using one electron redox chemistry combined with the rich Lewis acid catalysis inherent to Au complexes.










