Reports: ND455838-ND4: Enantioselective Hydrogen Atom Transfer Reactions from Transition-Metal Hydrides
 printer friendly
printer friendlySynthesis and Resolution of Chiral Ruthenium Complexes Containing the 1-Me-3-PhCp Ligand.
We were able to synthesize and resolve a set of half-sandwich ruthenium-complexes which are chiral because of the face-specific coordination of the 1-Me-3-PhCp ligand. This set includes complexes with acetonitrile, chloride and finally hydride ligands. They have been resolved into enantiomers via diastereomeric complexes with the (S)-a-methyl-benzenemethanethiolate ligand.
The synthesis of the racemic ruthenium chloride complex starts with refluxing the lithium salt of 1-Me-3-PhCp with tris(triphenylphosphine) ruthenium dichloride in ethanol. The bis(triphenylphosphine) complex obtained is treated with dppm in refluxing toluene, yielding the air-stable, racemic ruthenium complex 1. The whole synthesis is shown in Scheme 1.
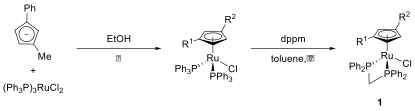
Scheme 1: Synthesis of racemic 1-Me-2-PhCp ruthenium complex 1 (R1 = Ph, R2 = Me or R1 = Me, R2 = Ph).
We have resolved racemic 1 into its enantiomers. As the cyclopentadiene does not allow the installation of a chiral auxiliary, we have installed a second chiral ligand on the Ru. For example, we prepared the diastereomers of 2 (Scheme 3) with an enantiopure nitrile ligand derived from mandelic acid.
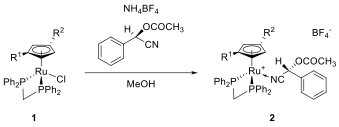
Scheme 2: Formation of diastereomeric complexes 2[BF4] with mandelonitrile-derivative. (R1 = Ph, R2 = Me or R1 = Me, R2 = Ph).
However, separation of the diastereomers of 2[BF4] by fractional crystallization failed. Therefore we pursued another pathway.
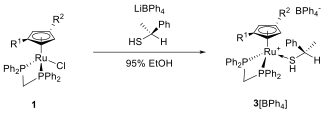
Scheme 3: Formation of 3[BPh4] upon treatment of 1 with thiol in presence of a Ph4- source.
We employed (S)-a-methyl-benzenemethanethiol as a chiral ligand. Treating 1 with the enantiopure thiol gave the complexes 3[BPh4] as a 1:1 mixture of diastereomers (Scheme 4). However, the diastereomers did not separate upon crystallization. The salt 3[BF4], obtained in an analogous synthesis, gave a glassy mass that was also unsuitable for fractional crystallization.
We removed the counterion by treatment with base, yielding neutral diastereomers of 4 (Scheme 4). These proved to be soluble in a variety of polar to nonpolar organic solvents. Vapor diffusion of pentane into a saturated solution of 4 in diethyl ether led to the growth of orange needles containing 92 % (SCp, R)-4, while the orange mother liquor contained 92 % of (RCp, R)-4.
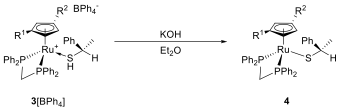
Scheme 4: Treatment of 3[BPh4] with base to remove the counterion.
The separated diastereomers were characterized by 1H, {1H}-31P NMR and X-Ray crystallography. The X-Ray analysis allowed unambiguous identification of the diastereomers. The S configuration of the thiolate ligand is of course preserved in both structures, while the configurations of the CpRu system are (as expected) oppposite. The phenyl substituent does not lie in the same plane as the Cp ring in either structure, the angle between the planes being 12.12° in (SCp, S)‑4 and 27.37° in (RCp, S)‑4.
Next we tried to replace the thiolate ligand by other ligands. Attempts to regenerate 1 with various chloride sources gave unsatisfactory yields, while attempts to replace the thiolate with acetonitrile failed or (at higher temperatures ) led to significant racemization.
Finally, we found that protonation of the thiolate in 3 with 1 equiv. of HCl or HBF4·OMe2 in Et2O to generate 3[Cl] or 3[BF4] was feasible. The thiol ligand we obtained was displaced easily by acetonitrile at room temperature without racemization. However, in the complexes we obtained, the acetonitrile ligand was too strongly bound to the Ru to be replaced by other ligands such as chloride or hydride. Treatment of 3[BF4] with LiCl in THF, in contrast, established an equilibrium between 3[BF4] and 1 (Scheme 5).
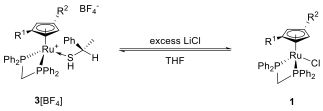
Scheme 5: Equilibrium between thiol complex 3[BF4] and chloride 1.
We found that the equilibrium lies substantially to the left, so it is impossible to isolate 1 from these mixtures. The addition of a drop of mercury provided a nice solution to that problem. By tightly binding the dissociated thiol mercury removed it from the solution, therefore driving the equilibrium all the way to the right and allowing the isolation of 1 in excellent yields after filtration through silica and removal of solvent. CD spectroscopy proved that no racemization had occurred during the ligand substitution.
Finally, we were able to synthesize the hydride complex (SCp)-5 by treatment of (SCp)-1 with sodium methanolate in methanol (Scheme 6). The retention of stereochemistry is complete, as shown by converting (SCp)-5 back to the thiol complex (SCp, S)-3[BPh4]; that diastereomer is the only one detected by {1H}-31P-NMR spectroscopy after the reaction.
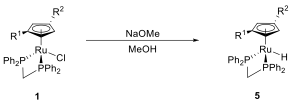
Scheme 6: Formation of the Ru hydride.
The PRF New Directions grant has enabled us to hire a postdoctoral associate from Germany (Tobias Dahmen) with experience in enantioselective chemistry. His expertise has been extremely helpful to my research group, and his experience with us has taught him how to think mechanistically about problems in catalysis.










