Reports: DNI653674-DNI6: Quantum Mechanical Calculations of Large-Scale Explicit Solvation
 printer friendly
printer friendlyThrough the support from the ACS-PRF Doctoral New Investigator grant in 2015-2016, we have performed investigations of how electronic excitation is treated with time-dependent DFT (TDDFT) in solution. In particular, we have combined quantum (TDDFT) and classical approaches to determine the best way to combine these methods for the best accuracy and computational affordability.
I. Convergence of Excitation Energies in Mixed Quantum and Classical Solvent: Comparison of Continuum and Point Charge Models
Mixed quantum mechanical (QM)/classical methods provide a computationally efficient approach to modeling both ground and excited state chemical processes in the condensed phase. To accurately model short-range interactions, some amount of the environment can be included in the QM region, while a classical model is used to treat long-range interactions to maintain computational affordability. The best computational protocol for these mixed QM/classical methods can be determined by comparison with converged molecular properties.
We compared molecular mechanical (MM) fixed point charges to a polarizable continuum model (PCM) for computing electronic excitations in solution. We computed the excitation energy of three pairs of neutral/anionic molecules in aqueous solvent, including up to 250 water molecules in the QM region. Interestingly, the convergence is similar for molecular mechanical point charges and a polarizable continuum model. For both classical models, large deviations (0.2-0.6 eV) in the excitation energy from converged values occur for the anions when no solvent is included in the QM region, but these errors decrease to ~0.1 eV if the nearest 5 water molecules are treated with QM (see Figure 1). The excitation energies of both neutral and anionic molecules are converged with respect to the number of QM water molecules when at least one full solvation shell is treated with QM.
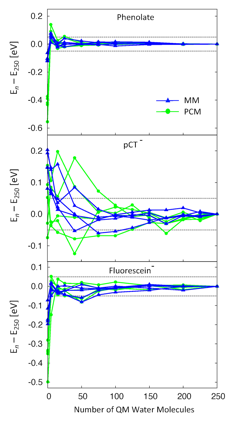
Figure 1. Deviation of QM/classical excitation energies from the values computed with 250 QM water molecules for five snapshots of phenolate (top), pCTÑ (middle), and the fluorescein anion (bottom). The classical region is modeled by TIP3P point charges (MM) or PCM with a SES cavity (PCM).
We found that while the van der Waals (VDW) definition of the PCM cavity is adequate for molecular structures with small amounts of QM solvent, larger QM solvent layers had gaps in the VDW PCM cavity, leading to asymptotically incorrect excitation energies (see Figures 2 and 3). Given that the VDW cavity leads to unphysical solute-solvent interactions, we advise instead using a solvent excluded surface cavity for QM/PCM calculations that include QM solvent.
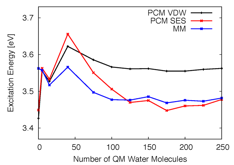
Figure 2. TDDFT excitation energies of a single snapshot of pCTÑ in water computed using a QM/classical approach where the classical region is modeled with TIP3P point charges (MM), PCM with a cavity defined by a VDW surface (PCM VDW), or PCM with a cavity defined by a SES (PCM SES).
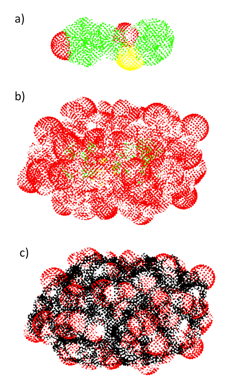
Figure 3. Tesserae of the PCM cavity surface of pCTÑ computed using a) VDW surface without QM solvent, b) VDW surface with QM solvent, and c) SES with QM solvent.
To test the effect of the magnitude of MM point charges on the convergence of QM/MM excitation energies, we computed excitation energies of pCTÑ using TIP3P point charges (oxygen = -0.8340e, hydrogen = 0.4170e), SPC/E point charges (oxygen = -0.8476e, hydrogen = 0.4238e), halved TIP3P point charges (oxygen = -0.4170e, hydrogen = 0.2085e), and doubled TIP3P point charges (oxygen = -1.6680e, hydrogen = 0.8340e). Results for a single snapshot of pCTÑ are shown in Figure 4. The QM/MM excitation energies computed with TIP3P point charges and SPC/E point charges agree within 0.01 eV for all amounts of explicit solvent. When the magnitudes of the TIP3P MM point charges are doubled or halved, the QM/MM excitation energies are noticeably different from those computed with standard TIP3P point charges. For example, the QM/MM excitation energies computed for one snapshot of pCTÑ using MM point charges that are half (double) the value of TIP3P point charges differ by up to 0.09 eV (0.21 eV) from values computed with TIP3P point charges. For all snapshots, the MM point charges with half (double) the value of TIP3P point charges require on average 150 (200) QM water molecules to converge the QM/MM excitation energy within 0.05 eV of the values computed with 400 QM water molecules, whereas TIP3P point charges require on average only 75 QM water molecules. Thus, varying the MM point charges did not lead to faster convergence, suggesting that tuning the charges will not lead to improved long-range results.
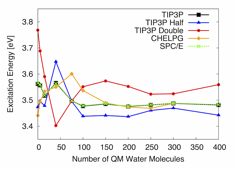
Figure 4. TDDFT excitation energies of a single snapshot of pCTÑ computed using a QM/classical approach where the classical region is modeled with TIP3P point charges (TIP3P), SPC/E point charges (SPC/E), half the value of TIP3P point charges (TIP3P Half), double the value of TIP3P point charges (TIP3P Double), and point charges derived from CHELPG analysis of 300 QM water molecules surrounded by ~4000 classical water molecules represented by TIP3P point charges (CHELPG).
Impact on career and training.
This project supported by ACS-PRF has allowed my group to continue generating results that will be used to apply for various junior faculty awards and future grants.
The funds have also been used to support 3rd year graduate Joel Milanese and post-doc Dr. Makenzie Provorse. Joel was able to concentrate on research full-time rather than having to balance research with teaching assistant duties, which has led to him being able to understand his project at a deeper level, and to be more productive with his calculations and analysis. Makenzie has made good research progress, has been able to mentor students, and plans to go on the academic job market. The funds also supported her travel to the ACS Philadelphia meeting to give a talk about this research.










