Reports: DNI654240-DNI6: Mesoscale Simulation and Machine Learning of Asphaltene Aggregation
 printer friendly
printer friendlySynopsis. Building on our results from the previous year, over the past 12 months of this award we have made four principal achievements. First, we have established a systematic protocol to construct coarse-grained models of asphaltene molecules from all-atom simulation data, and have used these models to perform the first molecularly detailed simulations of the complete asphaltene assembly hierarchy described by the Yen-Mullins model. Our findings are in good general agreement with the predictions of Yen-Mullins, providing theoretical support for a three-stage hierarchical aggregation model from monomers to nanoaggregates to clusters to a spanning network, but the details of which are strongly contingent on asphaltene chemistry and environmental conditions. These calculations have established new molecular-level characterization and understanding of multi-scale asphaltene aggregation as a function of concentration, temperature, and chemistry. Second, our mesoscale simulations reveal the kinetics of asphaltene association and dissociation at dynamic equilibrium to be well described by a Poisson process, with the lifetime of the rod-like nanoaggregates following an approximate exponential distribution. By fitting this phenomenological coagulation-fragmentation model to our mesoscale simulation trajectories, we have developed analytical models that enable us to understand and predict nanoaggregate lifetimes as a function of asphaltene chemistry, temperature, and concentration. Third, we have performed mesoscale simulations of aggregation as a function of temperature, pressure, and heptane/toluene solvent ratios to determine a phase diagram for assembly that enables prediction and engineering of aggregation state by controlling environmental conditions. Fourth, we have deployed nonlinear machine learning using a many-body adaptation of the diffusion map to discover and parameterize low-dimensional assembly landscapes representing the molecular-free energy surface for assembly as a function of cluster morphology.
Mesoscale model construction. We have established a general protocol to construct coarse-grained models of asphaltene molecules based on Boltzmann inversion re-parameterization of the Martini force field against all-atom calculations. We have constructed models for three prototypical continental asphaltene chemistries illustrated in Fig. 1, but the methodology is directly transferrable to arbitrary molecular architectures.
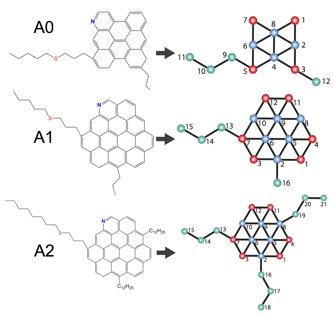
Fig. 1 – Coarse grained model development for three prototypical continental asphaltene architectures we term A0, A1, and A2.
Mesoscale assembly mechanisms. Our coarse-grained models permit us to conduct mesoscale simulations of hundreds of asphaltene molecules over microsecond time scales, leading to the first molecularly detailed simulations of the complete Yen-Mullins aggregation hierarchy (Figs. 2,3). Our results reveal aggregation to proceed by the formation of 1D rod-like aggregates of ~10 molecules driven by parallel stacking of the polyaromatic cores (Stage I), followed by the formation of clusters of ~6 nanoaggregates (Stage II), and finally the formation of a spanning network with fractal dimension of ~2 (Stage III). Interestingly, sufficiently long aliphatic side chains suppress the formation of nanoaggregate clusters due to steric repulsion, eliminating Stage II from the hierarchy.
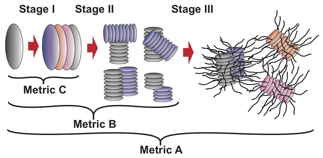
Fig. 2 – Schematic illustration of the Yen-Mullins assembly hierarchy observed in our mesoscale simulations of asphaltene aggregation.
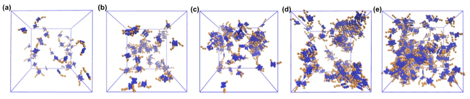
Fig. 3 – Representative snapshots of the aggregation hierarchy observed in mesoscale simulations of asphaltene A0 in n-heptane at 300K and mass fractions of (a) 5%, (b) 10%, (c) 15%, (d) 20%, and (e) 25%.
Kinetic model of nanoaggregate lifetime. Our mesoscale simulations reveal that nanoaggregate lifetime is well modeled by a Poisson coagulation-fragmentation process, where a particular 1D rod-like nanoaggregate "dies" either by binding with another rod or fragmenting into two daughter rods (Fig. 4). By fitting this model to our simulation data we have inferred rate constants for these processes as a function of concentration, temperature, and asphaltene architecture.
![]()
Fig. 4 – Poisson model for the lifetime distribution P(τN; λd(N)) of a nanoaggregate of N asphaltene molecules. τN is the lifetime, λd(N) the aggregation number-dependent death rate, λb the binding rate constant, λf the fragmentation rate constant, and Crods the nanoaggregate number concentration.
Phase diagram for assembly. By performing mesoscale simulations at various temperatures, pressures, and heptane/toluene solvent ratios, we have computationally determined an equilibrium phase diagram for aggregation (Fig. 5).
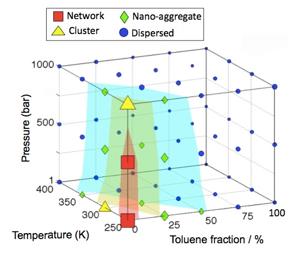
Fig. 5 – Aggregation phase diagram of asphaltene A0 predicted from mesoscale simulation.
Nonlinear machine learning of asphaltene assembly landscapes. We have applied diffusion maps to discover low-dimensional projections of mesoscale simulation trajectories into collective variables corresponding to the fundamental dynamical motions governing molecular-level aggregation (Fig. 6). These landscapes present a powerful framework with which to understand the thermodynamics, morphology, and kinetics of asphaltene aggregation, and reveal how environmental conditions and asphaltene architecture confine assembly dynamics to different regions of this manifold.
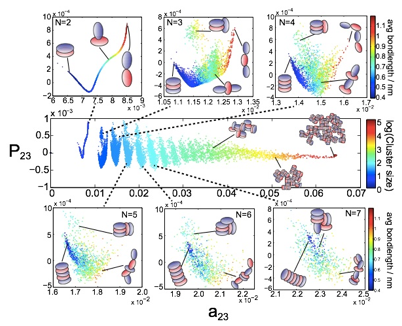
Fig. 6 – Two-dimensional assembly landscape for asphaltene A0. The two collective variables supporting the landscape (a23,P23) were discovered by nonlinear machine learning. The main figure shows the embedding of the variation of cluster size over the assembly landscape, and the insets the variation of the average distance between asphaltene molecules in the cluster.
Professional development. This award has permitted the PI to establish himself as a new investigator within the field of asphaltene modeling and self-assembly, allowing him to apply his expertise in molecular simulation and machine learning into a new arena to help understand and control asphaltene aggregation. The graduate researcher, Jiang Wang, supported by this award has benefited from close mentorship and training from the PI in molecular simulation, machine learning, and statistical thermodynamics, and in scientific writing and presentation. Jiang was the first author on a scientific publication resulting from this work published in J. Phys. Chem. B and also presented this work in Session C36: Soft Colloids: From Single Particle Properties to Bulk Phase Behavior and Dynamics at the 2016 APS March Meeting. The undergraduate researcher, Mohit Gayatri, who was supported on a 10-week research internship over Summer 2016 benefitted from the opportunity to gain first hand research experience, develop skills in molecular simulation and visualization, and preparation for graduate school. Finally, researchers at Aalto University, recently contacted us after reading our paper, and we are currently in discussions to explore the possibility of a computational/experimental collaboration.










