Reports: DNI354226-DNI3: Homogeneous Bimetallic Systems for Syngas Conversion
 printer friendly
printer friendlyThe proposed research focuses on the design of a new series of homogeneous bimetallic catalyst systems for conversion of syngas to value added chemicals and fuels such as alkanes, alkenes, and methanol. Within the context of this research we were interested in exploring related chemical transformations including CO2 reduction and H2 activation and evolution. This year our work primarily centered on these latter goals based on some exciting discoveries made while moving scientifically towards syngas conversion.
Last year we reported on the synthesis of a novel bimetallic complex with an eye towards exploring cooperative reactivity between the two metal centers (Scheme 1).

Scheme 1. Dimetallation of the bisimidazole phosphine ligand.
One major limitation of the above scheme is the fact that it generates a bimetallic complex bearing a coordinatively saturated RuCp* fragment in one of the ligand binding pockets. This motif precludes exploring cooperative reactivity between the two metal centers. As such we began to explore methods to metallate the starting bisimidazole phosphine ligand with Ru(II) precursors that would allow an open (or readily exposed) coordination site at ruthenium in the final complex. Ultimately we discovered that metalation with [Ru(Bz)(bpy)(OTf)]_ (Bz = benzene, bpy = bipyridine, OTf = triflate) in refluxing ethylene glycol gave us access to the desired dicationic biscarbene-phosphine complex with a bpy and an aquo ligand in the other three coordination sites ([1-OH2]2+). This molecule was structurally characterized as the monocationic chloride complex ([1-Cl]+) following addition of excess ammonium chloride to a solution of the complex (Figure 1).

Figure 1. Single crystal X-ray diffraction structure of [1-Cl]+ with thermal ellipsoids at the 50% probability level. H atoms are omitted for clarity.
When attempting to access the bimetallic complex using acetonitrile as a solvent and 2 equivalents of n-BuLi as a base in the presence of cobalt chloride, we discovered that instead of giving the Ru-Co bimetallic complex, as expected, an acetonitrile activated monometallic complex was obtained, demonstrating cooperative chemistry between the metal center and the ligand (Figure 2).
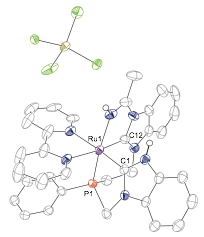
Figure 2. Single crystal X-ray diffraction structure of acetonitrile-activated complex with thermal ellipsoids at the 50% probability level. H atoms are omitted for clarity.
This unexpected result prompted us to explore the reactivity of this complex with other unsaturated substrates, including CO2 and bicarbonate. Addition of CO2 to solutions of [1-OH2]2+ in CH2Cl2 yielded orange crystals after standing at room temperature for several days. These crystals were analyzed by X-ray diffraction and were found to be a formate-bridged bimetallic complex (complex 2, Figure 3). Complex 2 was also independently prepared by the addition of sodium formate to a solution of [1-OH2]2+ in CH2Cl2. Direct addition of NaHCO3 to solutions of [1-OH2]2+ in CH2Cl2 also leads to complex 2 in low yield, consistent with the intermediacy of a Ru_HCO3_ species. Although other examples exist of reduction of HCO3_ to formate, they require added energy by initial formation of a M_H bond, the use of high pressures of H2, or the use of an applied electrochemical potential. Importantly, the formation of 2 from CO2 and HCO3_ represents a formal reduction of the added carbon species.
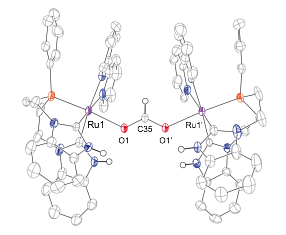
Figure 3. Single crystal X-ray diffraction structure of 2 with thermal ellipsoids at the 50% probability level. H atoms and counter ions are omitted for clarity.
In solvents in which 2 is soluble, the reaction proceeds further with conversion into a carbamate species, 3, with loss of H2 (Figure 4). The overall set of observed transformations is summarized in Scheme 2. These results are significant, as they represent a spontaneous reduction of CO2 without high-pressure H2, electrochemical potential, or strong reducing agents. We are continuing to study the initial reduction reaction to better understand this mechanism and its potential to operate catalytically for reduced carbon products. Scheme 2 may also suggest a pathway to store H2 if formal addition of H2O to 3 can generate a Ru_bicarbonate complex. This overall scheme simplifies to a water-splitting reaction that is catalytic in [1-OH2]2+ and CO2 (H2O + E _ O=E+H2) and provides a unique method of H2 production from water.
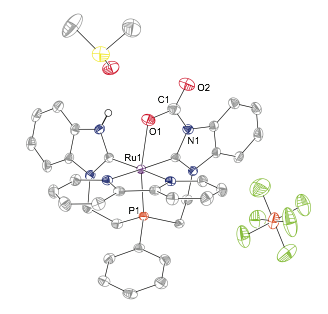
Figure 4. Single crystal X-ray diffraction structure of 3 with thermal ellipsoids at the 50% probability level. H atoms are omitted for clarity.
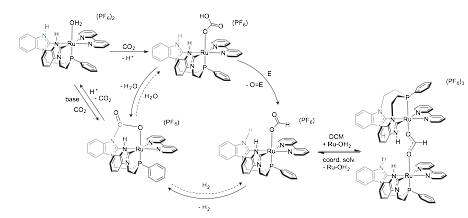
Scheme 2. Proposed Overall Transformation of Reduction of H2O to H2.
Moving forward we are interested in contrasting this reactivity at a single metal center to what may be achieved when a second metal replaces the carbene N-H moieties. We are excited at the prospect of achieving alternate modes of CO2 and H2 activation and ultimately conversion of C1 feedstocks, like syngas, into value added products.
Ultimately the work supported by this PRF award has helped to catalyze a whole new area of investigation in my lab focused on exploring metal-ligand cooperativity and ways a single ligand scaffold can be coaxed into varying modes of reactivity by design. We are excited about the future of this project and the science it will unlock in the realm of transformation of C1 feedstocks. The two primary students trained under this grant are expected to graduate next year and will go on to build on this foundation created during their PRF funding. One postdoctoral scholar who contributed to work on this project recently moved on to an independent faculty position at the University of Richmond.










