Reports: UR355304-UR3: Stoichiometric and Catalytic Nitrene-Group-Transfer Reactions from Late-Metal Silylamides
 printer friendly
printer friendlyIn this project, my research group is investigating the possibility that late-metal silylamides, particularly those of base metals, can serve as precursors to reactivity imido or related species that will participate in nitrene-group transfer. One goal of this work is to provide insights into how oxidative deprotection can be used as a strategy for unmasking reactive nitrogen groups in the presence of an appropriate metal to promote the reaction. A proposed catalytic cycle is shown in Figure 1, and we are particularly excited about the possibilities afforded by one-electron oxidation/deprotection steps to facilitate this reactivity at base metals, potentially in an electrocatalytic fashion.
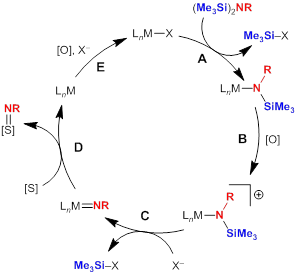
Figure 1. Possible catalytic cycle utilizing late-metal silylamides as protected nitrene-group sources
Project 1: Studies of (dtbpe)Ni Silylamides and their Redox Properties
Given the unique role demonstrated by Hillhouse for bis(phosphine) nickel complexes in supporting and transferring imido substituents to alkenes, we have initially targeted nickel silylamides supported by the 1,2-bis(di-tert-butylphosphino)ethane (dtbpe) ligand for exploration of steps B and C in the cycle from Figure 1. The silylamide complexes were prepared in high yield from the previously reported bis-μ-chloride dimer (Figure 2), and crystal structures have been obtained for two amides.
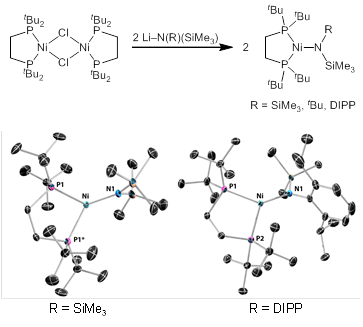
Figure 2. Synthesis and structures of (dtbpe)Ni silylamides
We initially proposed that introduction of silyl groups to nickel amides would increase electrochemical stability relative to the hydrogen substituents utilized by Hillhouse. This proposal was supported by cyclic voltammetry studies of all three silylamides (R = silyl, alkyl, and aryl), which all show a reversible NiI/II couple without the additional irreversible process observed for hydrogen-substituted amides (see Figure 3 for comparison of 2,6-diisopropylphenyl (DIPP) amido complexes). This electrochemical behavior supports the notion that the deleterious process previously reported is due to a proton-coupled degradation and suggests that silylamides are promising targets for the proposed catalysis. Moreover, the anodic shift observed relative to hydrogen-substituted analogues shows that the silylamido complexes may be much more reactive toward deprotection following oxidation.
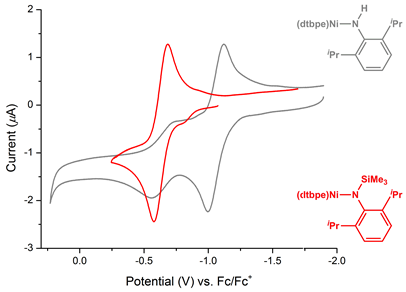
Figure 3. Voltammograms of (dtbpe)Ni amides with varying substitution
We have translated our electrochemical results into chemical redox, showing that the (trimethylsilyl)(DIPP) amido complex can be cleanly oxidized to the corresponding Ni(II) cationic silylamide. Consistent with electrochemistry results, the oxidation can be performed by a variety of chemical oxidants (unlike the Hillhouse system, which requires tropylium). We have found ferrocenium to react most cleanly and reproducibly. Though crystal structures are not yet available, spectroscopic evidence (particularly NMR) supports the assignment and suggests an unusual geometry whereby the amide is coplanar with the (dtbpe)Ni fragment. Based on comparison with related studies of (dtbpe)Rh amides in our lab, we believe that the difference is primarily due to the sterics associated with the DIPP substituent. Studies toward deprotection to form the known (dtbpe)Ni=N(DIPP) are ongoing (Figure 4).
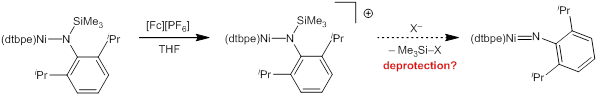
Figure 4. Oxidation of (dtbpe)Ni–N(TMS)(DIPP) and proposed deprotection
Project 2: Reactivity of (nacnac)Co Complexes
More recently, we have also devoted efforts to preparing cobalt complexes supported by β‑diketiminate (“nacnac”) frameworks, since such complexes have been shown to support terminal and bridging imido complexes. One target reaction has been to use O2 as an oxidant, wherein we would initially form a known Co oxo complex, which could be transformed into a Co imide through imide-for-oxo metathesis (Figure 5). We have prepared the known bis-μ-oxo precursor according to procedures from Warren. Although our initial attempts at imide synthesis from a disilylamine were unsuccessful, likely due to sterics, we have shown a reaction with chlorotrimethylsilane, which suggests that formation of a strong Si–O bond can in principle lead to Si–X cleavage at Co(III). Future efforts in this area will be devoted to understanding the synthesis and reactivity of a variety of low-valent cobalt complexes, including those supported by dtbpe, to understand differences in reactivity between Co and Ni.
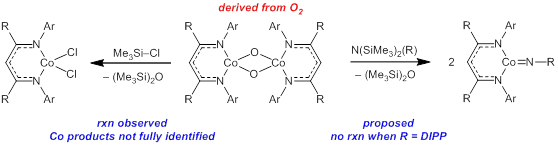
Figure 5. Proposed and observed reactivity for O2-derived [(nacnac)CoO]2










