Reports: ND655026-ND6: Understanding the Relationship between Fluid Molecular Structure and Pressure-Viscosity Behavior
 printer friendly
printer friendlyThis project is focused on understanding the connection between the viscous properties of lubricants and lubricant additives and the structure and chemistry of their constituent molecules. During this reporting period, we have explored two important properties: the increase of viscosity with pressure and the decrease of viscosity with temperature. The viscosity of a lubricant can change dramatically as a load is applied or the temperature rises during operation of moving components. Characterizing these changes is critically important to performance of lubricated moving components since frictional losses (and in turn energy efficiency) are directly tied to the viscosity of the lubricating fluid. Our goal is to fundamentally understand how the structure and conformation of fluid molecules change in response to pressure and temperature and how this determines viscous properties. The ultimate goal is to be able to predict the rheological properties of a fluid based only its molecular structure. In the following, we will summarize our progress in this first year of the project specifically related to the pressure-viscosity response of base fluids and the pressure-temperature behavior of base fluids with additives.
Typically, measuring the pressure-viscosity response of a fluid requires highly accurate experiments to be performed for each fluid composition. Model-based approaches would be preferred, but so far have been limited to two approaches. Atomistic models, such as Molecular Dynamics (MD) simulation, provide explicit representation of molecular structure as well as chemistry and can be used to predict pressure-viscosity behavior for nanoscale volumes of fluid. Empirical models, on the other hand, are mathematical equations developed from experimental observations that relate the rate of increase of viscosity with pressure to other material-specific properties. However, there are pros and cons to both approaches. MD simulations have structural precision that capture specific features of fluid molecules, but direct viscosity predictions demand large simulation sizes and long computational time, especially for complex fluids. Empirical models, on the other hand, are usually simple and straightforward mathematical equations, but they rely on experimental data and do not provide any insight on the dependence of molecular structure to pressure-viscosity behavior. To capitalize on the advantages of both, we developed a method in which ambient viscosity and pressure-volume data, which can be relatively easily obtained from MD simulations, is used with an empirical viscosity correlation to predict piezoviscous behavior of liquids.
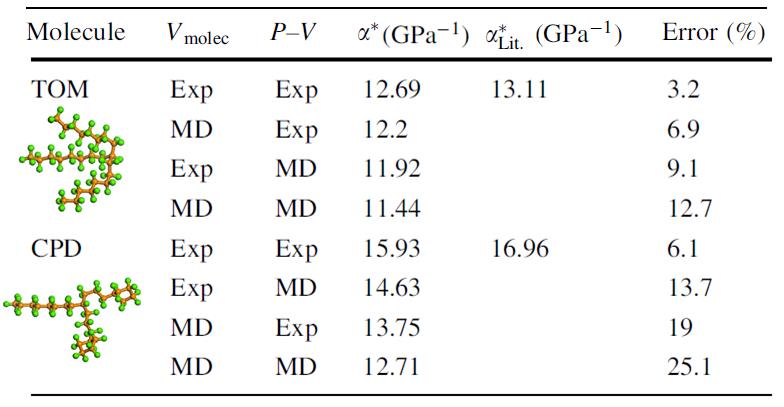
We calculated the asymptotic isoviscous pressure coefficient, alpha*, for several test fluids to evaluate this approach. Illustrative results are shown for 9-N-octylheptadecane (TOM) and 1-cyclopentyl-4(3-cyclopentylpropyl) dodecane (CPD) in Table 1. We observe that, while trends are captures, the predicted values may not be quantitatively accurate. This suggests that further research will be required to enable this method to be applied in practice. However, it is also evident that this method is quite promising. If the method can accurately predict the pressure-viscosity response for known lubricants, it could then be applied to make predictions for molecules with new or novel chemical structures, therefore enabling molecular-scale lubricant design. Further, the proposed approach suggests a means of fundamentally understanding the relationship between a fluid’s molecular structure and its pressure-viscosity behavior.
Table 1: Predicted pressure-viscosity coefficient and the resulting error in those predictions for TOM and CPD using pressure-volume (P-V) and molecular volume (Vmolec) data from either experiment or MD simulation.
Viscosity is also very sensitive to temperature and in most lubricants the rapid decrease of viscosity with temperature is mitigated with the use of Viscosity index (VI) improvers. These additives are typically high molecular weight polymers added is small volume fractions to the base fluid. The mechanisms behind the functionality of VI additives are still poorly understood. A widely accepted theory is the coil expansion mechanism, in which it is proposed that at lower temperatures the polymer is poorly soluble in the lubricating oil, tends to stay in a coiled conformation, and does not contribute much to fluid viscosity; then, at elevated temperatures, the solubility of the polymer in the lubricating oil improves, the polymer expands and induces a thickening effect on the solution, therefore reducing the decline of fluid viscosity with temperature. While this idea is widely accepted in the literature, there is little direct evidence to support it as a generally-applicable mechanism. In this study, we explored trends in polymer coil size using MD simulation.
The simulations were used to observe and characterize temperature-induced changes in the radius of gyration of model VI polymers in a base oil. Specifically, two model VI additives, random ethylene-propylene copolymer (OCP) and polydodecylmethacrylate (PMA), both with 50 repeat units, were placed in a dodecane solvent. A representative snapshot from one simulation is shown in the Table of Contents graphic. The radius of gyration of the polymers was calculated from simulations at 40 and 100 degrees C (reference temperatures for VI) as a function of time and from that data histograms of size data were generated, as shown in Figure 1. The simulations predicted that PMA will increase in size with temperature, while OCP will not. To understand the difference in coil size trends of PMA and OCP, we analyzed the structural and chemical disparities between these two molecules using simulations of test polymers to isolate the effects of specific differences. These simulations revealed that the presence of oxygen in PMA is important to the observed temperature-induced increase in coil size. Overall, the MD simulation method presented in this study holds significant promise since the ability to anticipate the effect of molecular structure and chemistry on coil size may enable molecular-scale design and optimization of novel lubricants.
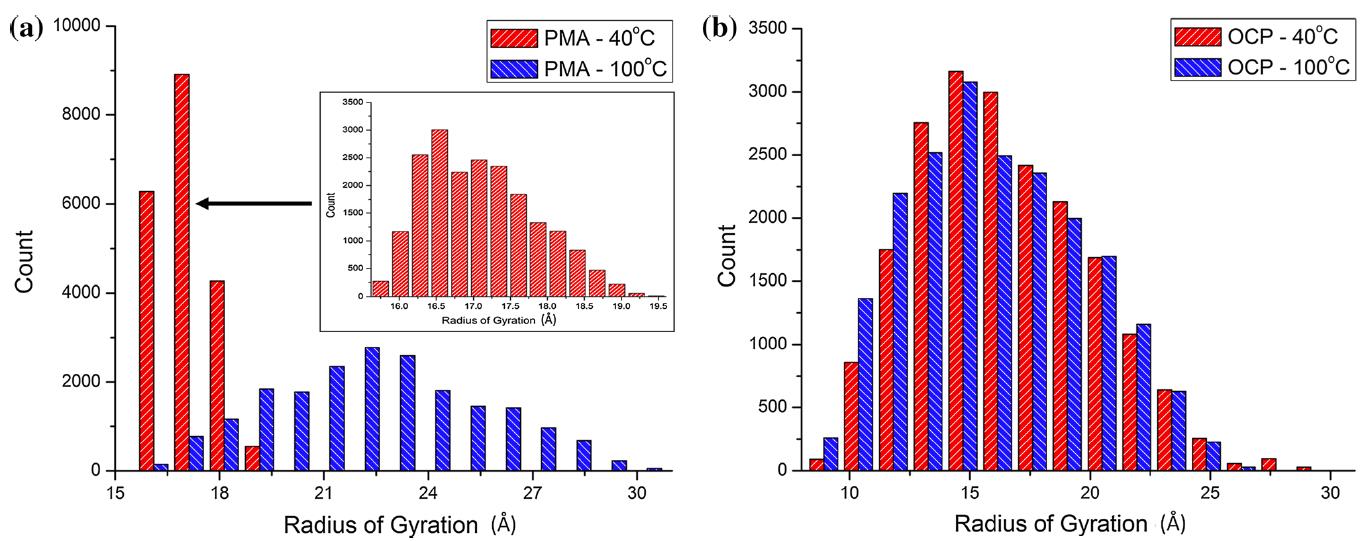
Figure 1: Frequency histograms for (a) PMA and (b) OCP at 40 and 100 degrees C. Gaussian functions are fit to these histograms to quantify the mean and standard deviation of the distribution
References:
U.S. Ramasamy, S. Bair and A. Martini, Predicting Pressure–Viscosity Behavior from Ambient Viscosity and Compressibility: Challenges and Opportunities, Tribol Lett (2015) 57:11
U.S. Ramasamy, S. Lichter and A. Martini, Effect of Molecular-Scale Features on the Polymer Coil Size of Model Viscosity Index Improvers, Tribol Lett (2016) 62:23










