Reports: UR353427-UR3: Homogeneous Fischer-Tropsch Catalysts for the Conversion of Syngas into Higher Order Hydrocarbons
 printer friendly
printer friendlyThe predicted decrease in the availability of petroleum and petroleum-based products has sparked renewed interest into the conversion of synthesis gas (syngas) into chemical feedstocks and fuels. Conversion of syngas (CO + H2) to liquid hydrocarbons and a-olefins by the Fischer-Tropsch (F-T) reaction offers promise, but the low selectivity of these reactions by the heterogeneous route is a major drawback. The development of homogeneous systems for the F-T reaction has the potential to increase both selectivity and understanding of the F-T mechanism. Funded by an Undergraduate Research grant from the American Chemical Society Petroleum Research Fund (UR, June 2013 – August 2016), metal-ligand complexes composed of pendant Lewis/Bronsted acids/bases and redox-active sites within the ligand scaffold are being investigated to study the C-H and C-C bond forming steps in the F-T reactions of CO.
Progress over the period of the grant included four major accomplishments outlined below. The first was to investigate utilizing the secondary coordination sphere to stabilize the formation of CO-derived iron formyls (formed from the addition of hydride to the reduced dicarbonyl species). To avoid deprotonation of the –CH3 groups on the 2,6-diacetylpyrindine core (Figure 1, left), which occurs upon the addition of hydride sources to the reduced carbonyl compounds (due to the acidity of the acetyl protons in these complexes) we synthesized complexes based on the 2,6-dibenzoylpyridine core instead (Figure 1). We continued to explore the reduction chemistry of these compounds by producing reduced iron dicarbonyls.
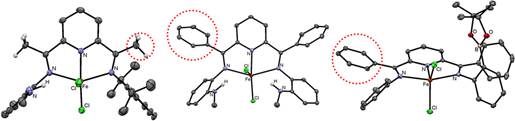
Figure 1. ORTEPs of representative FePDI complexes highlighting the acidic –CH3 (left) and the nonacidic –C6H5 groups (middle and right) in the PDI backbone.
Unfortunately we were not able to observe formyl formation upon addition of hydride sources to the reduced dicarbonyl analogs.
Second, we published an in-depth study of the relationship between protonation state of the secondary coordination sphere pendant base and the redox-activity of the PDI ligand scaffold. Given the intimate nature of secondary coordination sphere protonation state and redox potentials in catalytic systems, we also reported the pH dependence of the potentials of the redox-active ligand in the reduced FePDI complexes. The protonated didpa ligand is capable of forming metal halogen hydrogen bonds (MHHBs) in neutral complexes and the solution behavior of the MHHBs was probed via pKa measurements and 1H NMR titrations of H-bond accepting strength. The relationship between the protonation state and the ligand-based redox activity was also probed (Figure 2) utilizing where the reduction potential of the didpa scaffold was found to shift by 105 mV upon protonation of the reduced ligand in Fe(didpa)(CO)2. That work was published as an article in Inorganic Chemistry.
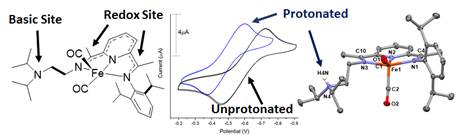
Figure 2. Image outlining the relationship between the redox potential of the reduced Fe(didpa)(CO)2 complex and the protonation state of the ligand. The reduction potential of the complex shifts by ~100 mV upon protonation/deprotonation of the ligand.
Third, in continuing with the theme of novel metal-ligand complexes based on the pyridinediimine (PDI) scaffold, we successfully synthesized and characterized a series of complexes with pendant redox-inactive metals located in the secondary coordination sphere (Figure 3). Redox-inactive metals are often used in combination with redox-active transition metals in synthetic and biological systems to invoke reactivity involving electron transfer. The reduced dicarbonyl complex Fe(15c5PDI)(CO)2 was shown in both the solid state and solution to encapsulate redox-inactive metal ions, where the reduction potential of the metal-ligand scaffold was found to shift by ~50 mV upon encapsulation of either Na+ or Li+. That work was published as a communication in Inorganic Chemistry.
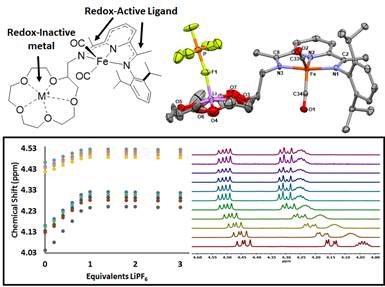
Figure 3. Graphic describing the structure of [Fe(15c5PDI)(CO)2Li][PF6] (top) and solution 1H NMR titration of Fe(15c5PDI)(CO)2 with LiPF6 in acetonitrile-d3.
The fourth area of study was characterizing the electronic structure of rare and unusual copper(I) complexes. The geometry of copper complexes is intimately linked to properties such as electron transfer rate, as typified by the family of blue copper proteins (BCPs). The fast electron transfer rates in BCPs are due to imposed geometric constraints on the copper centers known as the “rack” or “entatic” states. In most copper complexes, Cu(II) can be found in distorted octahedral, square pyramidal, or trigonal bipyramidal coordination, while Cu(I) almost exclusively adopts a tetrahedral geometry. Cu(I) complexes that exhibit square planar geometry are rare, and in most cases are stabilized in multimetallic systems. Utilizing the sterically bulky tridentate pyridinediimine ligand, iPrPDI (where iPrPDI = 2,6-(2,6-iPr2C6H3N=CMe)2C5H3N), we reported a series of monometallic square planar Cu(I) complexes (Figure 4). The manuscript was published in Chemical Communications.
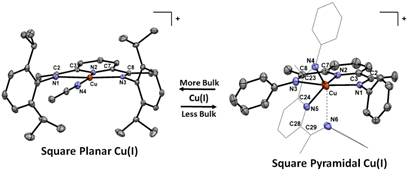
Figure 4. Graphic illustrating the square planar copper(I) compound [iPrPDICu(I)(CH3CN)]+ (left) and square pyramidal copper(I) compound [PDI2Cu(I)]+ (right).
There
were two students that received summer stipends during the 15-16 reporting
period (Kyle Burns and Pui Man Cheung). Both
students graduated with a B.S. in Chemistry from WWU and both are pursuing a Master’s
degree in Chemistry at WWU. The grant
has also had a positive effect on the career of the PI, providing valuable data
and publications in pursuit of federal funding from agencies such as the NSF
and NIH.










