Reports: ND353962-ND3: Investigation of a Straightforward and Highly Customizable Synthetic Route to Triply Bidentate Phosphine Ligands
 printer friendly
printer friendlyLast year we reported on our successful synthesis of [1,1'-biphenyl]-2-yldimethylphosphine (Methyl-JohnPhos; MeJPhos for short; see Figures 1 and 2). JohnPhos ligands have long been valued for two design features, namely a strong σ-donating dialkyl-phosphine and a sterically demanding biphenyl moiety. Though JohnPhos-type ligands have been thoroughly studied for their properties by derivatization of the biphenyl moiety, very little effort has gone into determining effects of the alkyl groups bonded to phosphorus. Because only larger alkyl groups (tert-butyl, cyclo-hexyl, adamantyl, etc.) have been studied, it was not clear how small alkyl groups on the phosphine would affect metal complex reactivity. We hypothesized that a shorter phosphorus-metal bond distance for the smaller R-JohnPhos ligands could provide superior σ-donation while retaining enough steric hindrance at the biphenyl moiety (which is necessary for reductive eliminations in Pd cross-coupling reactions). Accordingly, the focus of our PRF-supported research this past year was to investigate the effects of the alkyl groups in a range of JohnPhos ligands.
To investigate the effect of the alkyl groups, we synthesized the JohnPhos ligands shown in Figure 1. Our specific goal was to determine how the various alkyl groups affect the electronic and structural properties of metal complexes containing these ligands. Note the o-biphenyl moiety is preserved throughout.
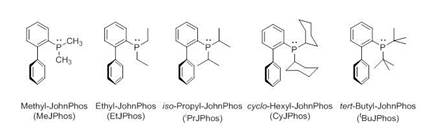
Figure 1. Structures, names, and abbreviated names of the R-JohnPhos ligands investigated in our study.
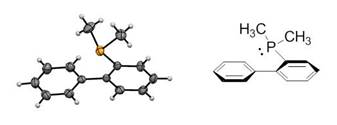
Figure 2. ORTEP crystal structure of MeJPhos (all H atoms refined). The biphenyl torsion angle is 66.8° and the ÐC-P-C bond angles are 100.3°, 102.7°, and 98.7°. Thermal ellipsoids are drawn at 50% probability.
The π-acidity and σ-donating properties of the R-JohnPhos ligands were quantified using a Cotton-Kraihanzel analysis on the Cr0(CO)5(R-JohnPhos) complexes. Very surprisingly, the s-donating ability of the R-JohnPhos ligands follow the trend tBu-JohnPhos < Et-JohnPhos < iPr-JohnPhos < Cy-JohnPhos << Me-JohnPhos. (The expected trend based solely on the s-donating ability of the R groups would be Me < Et < iPr ~ Cy < tBu.) To explain this trend, we analyzed the X‑ray crystallographic data of 22 metal complexes of the type trans-Cr0(CO)4(PR3)2, Pd0(PR3)2(η2-dba), and trans-PdII(Cl)2(PR3)2 (e.g., Figure 3).
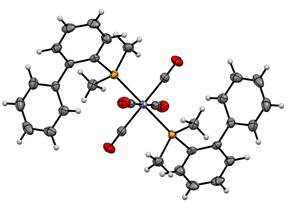
Figure 3. ORTEP crystal structure of trans-Cr0(CO)4(MeJPhos)2 (all H atoms refined). Thermal ellipsoids are drawn at 50% probability.
Specifically, the geometric distortion at the phosphorus atoms in these complexes was examined to determine the back-strain. (The distortion is directly analogous to back strain experienced by sterically crowded amines.) For phosphines, this geometric distortion is measured using the symmetric deformation coordinate called S4' (Figure 4). (The S4' value is the angular difference between the three ligand ÐC-P-C angles and ÐC-P-Pd angles.) The distortion from trigonal pyramidal at the phosphorus directly correlates to the hybridization of the lone pair. More distortion towards trigonal planar, caused by sterically bulky R groups at phosphorus, forces the lone pair (HOMO) into a p-like orbital that in turn has poorer overlap with the metal. This distortion occurs when strong steric influences at the phosphorus forces the geometry to be more planar, which results in near-zero or negative S4' values.
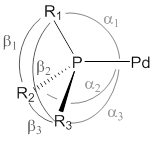
Figure 4. Structural and geometric parameters defining the symmetric deformation coordinate (S4'). S4' = (α1 + α2 + α3) - (β1 + β2 + β3).
Our analysis showed that the R-JohnPhos ligands are exceptionally sensitive to back strain compared to typical phosphines. These results suggest that the strong s-donating ability of the Methyl-JohnPhos ligand can be attributed to its ability to avoid both back strain and front strain. Consequently, the -PMe2 moiety allows for very short phosphorus-metal bond distances. Overall, the observed ordering of the s-donating ability for the R-JohnPhos ligands (tBu-JohnPhos < Et-JohnPhos < iPr-JohnPhos < Cy-JohnPhos << Me-JohnPhos) was proposed by us to arise from competition between the intrinsic electron-donating ability of the R groups (Me < Et < iPr ~ Cy < tBu) and the steric interactions (front and back strain) that decrease the electron-donating ability of the phosphine.
Finally, it is important to note that, because of the sterically dominating o-biphenyl and close phosphorus-metal bond distances, MeJPhos maintains a large overall steric profile that is actually larger than CyJPhos, as measured by percent buried volume (%Vbur). This is important because it suggests that Me-JohnPhos will be able to function as a ligand in catalytic cross-coupling reactions. Overall, we concluded that the -PMe2 moiety is a powerful way to incorporate strong σ-donation into “designer” phosphines while retaining other advantageous structural (e.g., size) and reactivity properties.
In work that we have not yet published, we developed a bench-top method for the synthesis of heteroleptic alkyl-phosphine oxides and heteroleptic alkyl-phosphines. Prior to the development of our method, there was no good route to the synthesis of these materials. Most researchers use a route reported by Hays (Figure 5). This method is a two-step, one-pot reaction with poor yields, egregious workup conditions, and very limited substrates. Of most concern is the sensitivity of this reaction to unusual stoichiometry (requiring a wasteful excess of alkyl bromide) and the vulnerability to considerable side reactions. These factors make this synthetic route intractable. Although the Hays’ method has been successfully applied to a narrow scope of phosphine syntheses, as reported by Doyle, it is basically an unworkable method for the preparation of heteroleptic phosphines oxides.
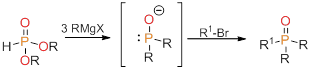
Figure 5. The Hays method of tertiary phosphine synthesis.
Our modified route is shown in Figure 6. The result of this methodology is a simple benchtop synthesis of tertiary phosphine oxides that is exceptionally amenable to complex alkyl halide precursors. The synthetic usefulness of this method has been briefly touched on with the synthesis of unsymmetrical bis(phosphine oxides); however, the simplicity in the synthesis makes this applicable to many more complicated systems (such as tri- or tetra-dentate phosphines, chiral SPOs and/or chiral electrophiles, etc). We are currently investigating the mechanism of the phosphinite anion attack on electrophiles in more detail as well as developing a library of new unsymmetrical phosphines using this method.
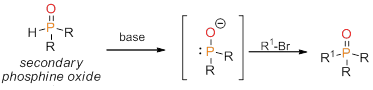
Figure 6. Our modified route to heteroleptic phosphine oxides.










