Reports: DNI654235-DNI6: Molecular Modeling of Shape Transformations in Polystyrene Vesicles
 printer friendly
printer friendlyThe overall goal of this proposal is to develop new coarse-grained models of polystyrene and polystyrene-based membranes to characterize shape transformations in polystyrene vesicles. To begin with, the PI and her laboratory have mostly completed proposed research as proposed in Objective 1, but have not yet completed aims as proposed in Objective 2.
Objective 1. i) Develop experimentally accurate coarse-grained molecular models for PS and small molecular additives such as THF. ii) Extend the model to study glassy PEO-PS membranes and investigate copolymer composition effects on membrane properties.
In order to develop experimentally accurate coarse grained molecular models for PS and extend this model to study glassy PEO-PS membranes the PI and her laboratory first characterized the hydrophobic collapse of single polystryene chains in water using molecular dynamics simulations.
We have designed a coarse grain model for PS based on all-atomistic simulations. This model based on the specific molecular chemistry of polystyrene is shown in Figure 1a, maintaining an extremely close mapping with the center of mass of the backbone and the phenyl groups. We map the phenyl rings onto three CG beads, while we map the backbone beads onto a bead that is centered between the two backbone carbons. Differing from both the A and B models of Rossi et al, this model includes a backbone bead centered between either Cb-Ca-Cb, or Ca-Cb-Ca and remains structurally more consistent with the underlying polystyrene structure.
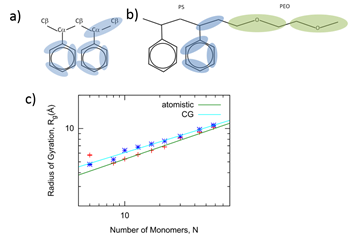
Figure 1. a) Coarse grained (CG’ed) mapping proposed for new PS model, similar to the model by Rossi et al., but the backbone mapping includes a bead centered between the two carbons. b) CG’ed mapping for new PEO-PS model. c) Initial results confirm scaling of Rg vs. N for CG’ed model, as compared with all-atomistic simulations.
Scaling Behavior of Single PS Chains. To begin with, we have tested the PS model in bad solvent environments (in water) to ensure the proper scaling behavior, in comparison with results from all-atomistic simulations. According to Flory, we can write the free energy of a polymer chain as a sum of the elasticity of the chain and the internal energy. Summing the elastic and internal energy contributions to the free energy and then taking the appropriate limits for strong swelling or strong compression, it is found that Rg ~ Nν, where ν is the scaling exponent. For a good solvent, ν = 3/5 and the polymer behaves like an extended coil. In a bad solvent with ν = 1/3, the polymer collapses from a coil to a globule to minimize contact with the solvent. For an ideal θ solvent with ν = 1/2, the polymer chain dimensions scale like a random walk. By fitting <Rg> ~ Nν as shown in Figure 1c, we find the average radius of gyration for polymer chain at the minimum energy state, from metadynamics calculations of a range of all-atomistic polystyrene lengths from 5 to 58 monomers: Rg(N)= 2.31 N0.36, providing a scaling coefficient ν=0.36 ± 0.02199 close to 1/3, expected for collapsed states of long hydrophobic polymer chains. Metadynamics is a methodology whereby a Gaussian potential is applied with time to the free energy surface until it can overcome free energy minima. Metadynamics has been extensively applied to accurate sampling of the free energy surfaces of protein configurations, chemical reaction pathway, and other phenomena. Similarly, by fitting <Rg> ~ Nν, we find that the average radius of gyration for our CG polymer chain model at the minimum energy state from metadynamics calculations is Rg(N) = 2.88 N0.32, with scaling coefficient ν = 0.32 ± 0.01865 close to 1/3. The close agreement between CG and all-atomistic scaling results provides initial validation for the PS model.
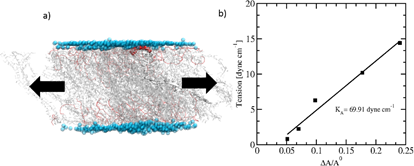
Figure 2. On left, a) snapshot after 100ns simulation of a CG’ed model of a PEO-PS diblock copolymer membrane, specifically PEO11-PS43. b) Artificially stretching the membrane laterally shows an increase in surface tension. The slope leads to an estimate of the area elastic modulus, KA, of approximately 70 dyne/cm.
Membrane Elasticity. Given similar values of Hildebrand solubility coefficients for PS and polybutadiene (PBD), we estimate that the interfacial tension between PEO and PS, gPEO-PS, will be similar to PEO and PBD, gPEO-PBD = 26 mN/m. From the interfacial tension value we can estimate the area elastic modulus, kA = 4g = 102 mN/m, which is independent of the molecular weight of the polymer. We can make a simple computational estimate of kA for a PEO-PS bilayer membrane from the slope of the tension (τ) vs. area expansion, A-A0/A0 (DA/A), for a given set of diblock lengths. In Figure 2a, on left, we show snapshot after 100 ns simulation of a CG model of a PEO-PS diblock copolymer membrane, specifically PEO11-PS43. Artificially stretching the membrane laterally shows an increase in surface tension. From the slope of surface tension vs. area expansion as shown in Figure 2b, we obtain an initial computational estimate of the area elastic modulus, kA, of approximately 70 dyne/cm or 70 mN/m, which is lower than the initial theoretical estimate of 102 mN/m.
Impact.
This grant has allowed for the partial support of two postdoctoral fellows. Additionally, one graduate student has worked on this project. She is in her final year as a CUNY Graduate Center Physics PhD. Her work on this project was recognized with a fellowship for her final year of research. Additional work on this grant has served as the basis for a recent NSF CAREER Award Proposal.










