Reports: ND353349-ND3: Well-Defined Zinc and Aluminum Catalysts for Reduction of Petroleum Derived Products
 printer friendly
printer friendlyIn this final stage of funding, our main efforts were concentrated on finishing subprojects of the original proposal.
1) Activation of sigma bonds on Al(I) center
We have extended our research on the activation of s bonds H-X (X = H, Si, B, Al, C, N, P, O) and C-X (X= O or F) by Al(I) compound NacNacAl (1, NacNac = [ArNC(Me)CHC(Me)NAr]− and Ar = 2,6-Pri2C6H3)[1] to the activation of element-element and carbon-element bonds. Thus, we found that 1 cleaves the S-S bond of diphenyl disulfide at room temperature to form the respective four-coordinate bis(phenyl sulfide) complex NacNacAl(SPh)2 (2). Oxidative addition of the C-S bond of diethyl sulfide to compound 1 takes place upon heating to 50 °C to furnish the thiolate aluminum complex NacNacAlEt(SEt) (3). The latter reaction is of interest in the context of developing new methods for desulfurization of petroleum products. A reaction of 1 with tetraphenyl diphosphine gives the bis(diphenyl phosphido) complex NacNacAl(PPh2)2 (4). Compounds 3 and 4 are the first examples of C(sp3)–S and R2P–PR2 bond activation by a main-group element complex. All three new compounds were characterized by multi-nuclear NMR spectroscopy and X-ray crystal structure analysis. Furthermore, we found that complex 4 is fluxional because of restricted rotation of the bulky PPh2 groups. This dynamic behaviour was studied by a variable-temperature NMR.
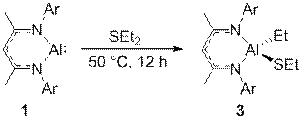
Scheme 1. Oxidative addition of the (sp3)C‒ S bond of diethyl sulfide to 1
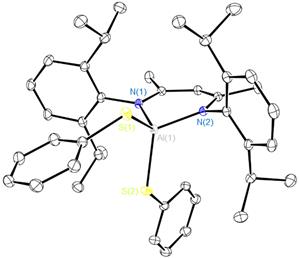
Figure 1. Molecular structure of 2.
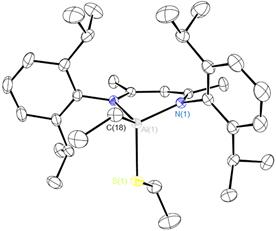
Figure 2. Molecular structure of 3.
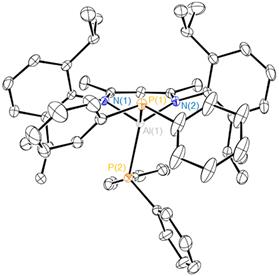
Figure 3. Molecular structure of 4.
2) Activation of double bonds on Al(I) center
This project emerged as a natural extension of the previous project. We noticed that the energy of the C=S bond (137 kcal mol–1) is not that much higher than the energy of C-F bond (116 kcal mol–1). Given the fact that the latter can be easily cleaved by complex 1 (see 2015 report for details), we thought that a similar oxidative cleavage of the C=S bond is possible. Oxidative addition of RnX=YRm to an Al(I) center is not possible because the reaction would be a four-electron process. However, we supposed that if either the RnX or RmY fragment could act as a two-electron donor to the resulting metal center, the oxidative addition of this multiple bond would be an allowed, two-electron process. We tested this hypothesis in the addition of cyclic thiourea 5 to compound 1 and indeed observed cleavage of the C=S bond and formation of the terminal sulfido complex 6 supported by a N-Heterocyclic carbene (NHC).
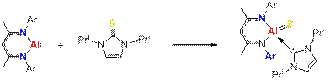
Scheme 2. Oxidative cleavage of the C=S bond on 1
The new compound 6 was characterized by spectroscopic methods and X-ray diffraction analysis (Figure 4).
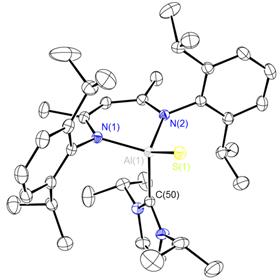
Figure 4. Molecular structure of 6.
Related reaction of 1 with 2 equivs of Ph3P=S (P=S bond energy is 80 kcal mol–1) results in an unstable, Ph3P=S-stabilized sulfido complex 7. Interestingly, at low temperatures (<−30 °C), compound 7 is fluxional due to facile dissociation of phosphine sulfide and its recoordination on the opposite face of the NacNacAl fragment (Scheme 3). Above −30 °C, it decomposes into dimer [NacNacAl(μ-S)]2.
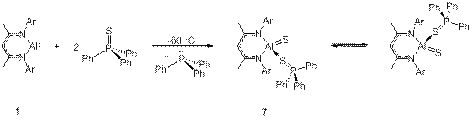
Scheme 3. Generation of 7 at low temperature from the reaction between 1 and triphenylphosphine sulphide and its fluxional behavior.
3) New neutral and anionic zinc compounds stabilized by a NCN pincer ligand.
The germanium and tin hydrides 8 have been previously reported by Roesky et al. but their reactivity has not been delineated yet.[2] We targeted their zinc analogue 9 because we reckoned that the increased polarity of the Zn-H bond will impart a greater reactivity to this species. Indeed, complex 9 could be cleanly generated by the reaction of the precursor 10 with L-Selectride but happened to be too unstable and quickly rearranged at room temperature to the amide species 11 (Scheme 4). NMR data for 11 showed a Cs symmetric pattern in solution, suggesting the presence of a tri-coordinate zinc compound 12. However, X-ray diffraction study from a crystal grown from this solution showed an unusual rearranged geometry, with one four-coordinate and one two-coordinate zinc atoms (Figure 1). We assume that this dimeric structure is in a fast equilibrium with the monomer 12. Interestingly, a reaction of 11 with THF and LiCl leads to an unusual hydride abstraction reaction and formation of the zincate species 13.
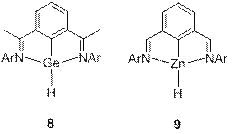
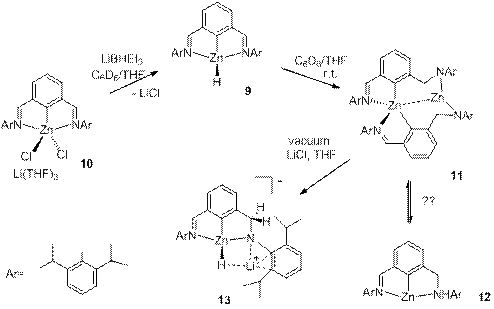
Scheme 4. Preparation of zinc pincer compounds
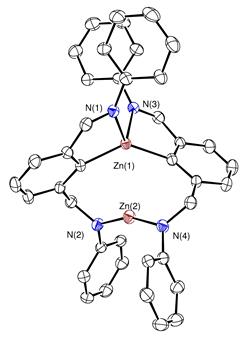
Figure 5. Molecular structure of 11
4) Hydrosilylation of olefins and alkynes by cationic aluminum complexes
The cationic aluminum complex [NacNacAlH]+ (14, NacNac = CH{C(Me)N(2,6-Pri2C6H3)}2) is isolobal to the zinc complex NacNacZnH, which was previously found to be a catalyst for chemoselective hydrosilylation of nitriles.[3] However, attempted application of 14 to hydrosilylation of hard substrates L (ketones, nitriles, imines etc) led to the formation of stable and catalytically inactive adducts [NacNacAl(L)H]+. However, complex 14 catalyzes hydrosilylation of olefins and alkynes by HSiEt3. Mechanistic studies showed that, although olefin insertion into the Al–H bond is very facile, the catalysis does not proceed by an insertion/metathesis mechanism but more likely by Lewis acid activation. Stoichiometric reactions of 2 with alkynes furnished unexpected products of C≡C addition across the NacNacAl moiety to give tripodal aluminium cations, which are also potent catalysts for the hydrosilylation of alkynes (Scheme 5).

Scheme 5. Preparation of tripodal compounds by addition ofalkyne to aluminum complex 14
[1] Chu, T. ; Korobkov, I.; Nikonov, G.I. J. Am. Chem. Soc. 2014, 136, 9195−9202.
[2] Khan, S.; Samuel, P. P.; Michel, R.; Dieterich, J. M.; Mata, R. A.; Demers, J.-P.; Lange, A.; Roesky, H. W.; Stalke, D. Chem. Commun., 2012, 48, 4890–4892.
[3] Boone, C.; Korobkov, I.; Nikonov, G. I. ACS Catal. 2013, 3, 2336-2340.










