Reports: DNI955467-DNI9: Nonaqueous Electrochemical Activation of Methane under Ambient Conditions for High-Value Liquid Fuel Production
 printer friendly
printer friendlyThe overarching goal of this project is to efficiently activate methane to higher value liquid fuel and chemicals. We propose to selectively activate C-H bonds in methane via a free radical cation-mediated activation pathway in nonaqueous media, which involves four key processes: 1) C-H bond activation in methane via an electro-oxidation reaction to produce methyl radicals and protons at the anode; 2) formation of higher hydrocarbons (C2-C12) via the chain growth mechanism initiated by the methyl radicals in the electrolyte; 3) transport of proton from anode side to cathode side in the nonaqueous medium; and 4) electro-reduction of proton to produce hydrogen at the cathode.
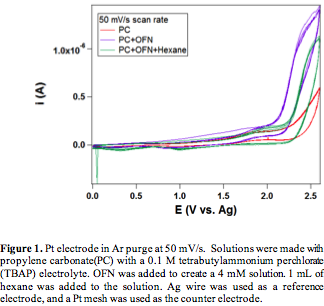
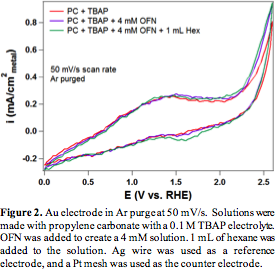
We started our investigations by replacing methane with hexane due to the low solubility of methane in the nonaqueous solvents. Octafluoronapthalene (OFN) is chosen as the radical initiator owing to its high redox potential. The cyclic voltammograms (CVs) of hexane in propylene carbonate (PC) on Pt show that the introduction of OFN leads to pronounced increase in current compared to the OFN free case (Figure 1). This is indicative of the redox cycle of OFN∙+/OFN. However, when hexane was added to the OFN containing electrolyte, the current decreases, which suggesting that the presence of hexane inhibits the redox cycle of OFN. We attribute this to the deactivation of the Pt electrode by carbon deposit from excessive C-H bond activation in hexane (evidenced by slide color change of the Pt electrode after reaction), most likely due to the ability of Pt to activate C-H bonds. Thus, we switch to Au, which is known for its inertness towards C-H bond activation. For the Au electrode, there is again an increase in the current response with the addition of the OFN. Further, adding hexane also increases the current response as desired (Figure 2). The increased current response is attributed to the reaction of hexane reacting with OFN_+, which is the desired reaction. The potential at which the current increased the most in the presence of hexane is at 2.4 V vs. Ag wire reference, which is likely the potential at which products are formed at the highest rate.
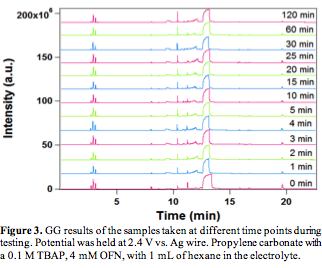
To accumulate enough products for gas chromatography (GC) and mass spectrometry (MS) analysis, we conducted long term potentiostatic tests with an Au foil with a surface area of 0.5 cm2 at 2.4 V vs. Ag wire for 2 hours. Small samples were taken from the reaction mixture and injected into a GC/MS at different reaction times (Figure 3). However, no appreciable change is observed during the reaction time, expect in the retention time range of 11-13 min. Detailed MS fragmentation analysis shows that this is due to the degradation of the TBAP to tributylamine as well as some OFN derivatives. No detectable level of dodecane or other larger hydrocarbons is observed in MS. Direct sampling of the headspace by MS is also attempted, but no desired product is detected. Since the C-H bond strength in methane is higher than hexane and the solubility of methane in PC is low, we consider it is unlikely that the proposed electrochemical approach will produce detectable levels of higher hydrocarbons. In addition, the high redox potential of the OFN_+/OFN pair is likely to slowly degrade the electrolyte and solvent, which also greatly complicatse the detection of small amounts of desired hydrocarbons. Therefore, in the interest of making concrete progress in methane activation within the funded period, we propose to explore a new approach: combining thermochemical cycle with dehydroaromatization (DHA) to efficiently and selectively convert methane to aromatics.
Nonoxidative dehydroaromatization (DHA) converts methane into aromatics, e.g., benzene, toluene and naphthalene, and hydrogen with a theoretical 100% atom efficiency toward valuable aromatic compounds:
![]()
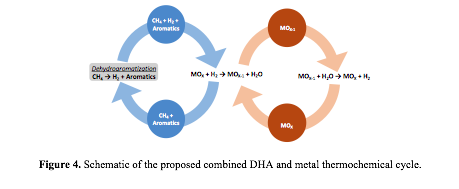
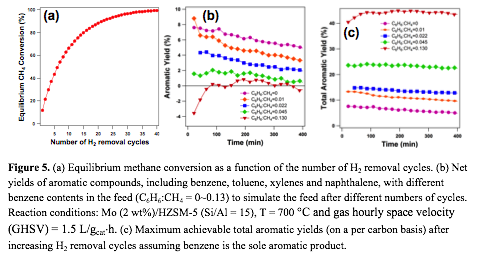
High selectivity for benzene (>80%) was reported on Mo supported HZSM-5 bifunctional catalysts. However, DHA of methane is severely limited by the thermodynamics, with an equilibrium conversion of methane at 700 °C below 15%. We propose to remove hydrogen produced in the DHA step by reacting the effluent with a metal oxide to form a hydrogen depleted feed for the next cycle of DHA. The reduced metal oxide can be recovered by reacting with water to form H2. Thus, the overall cycle (Figure 4) is equivalent to a reactive extraction process of hydrogen from the DHA system. Thermodynamic calculations show that the equilibrium conversion of methane increases steadily with the number of H2 removal cycles (Figure 5). We have finished building the experimental setup for evaluating both methane DHA and hydrogen removal steps. Mo-ZSM-5 catalysts have been synthesized and characterized. Similar activity and product distribution in methane DHA on Mo-ZSM-5 to those in the literature have been observed. Thus, we are currently evaluating the feasibility of the proposed DHA/thermochemical cycle by: 1) screening metal oxides to selectively remove H2 from the CH4/H2/aromatics mixture; and 2) elucidating the impact of benzene on the activity and product distribution of methane DHA in the absence of H2.
The proposed concept has the potential of developing an efficient and selective approach in converting methane to aromatics in a single reaction step. The combination of DHA and thermochemical cycle represents a novel reactive separation method in removing hydrogen from the reaction mixture, which can be reformed in a separate step. If implemented, this concept could significantly improve the economic viability of DHA.
The support from ACS PRF has been a key enabling factor in jump-starting my newly established research program at University of Delaware. We have explored two distinctive approaches in methane activation, which is a major challenge in the petroleum research field. Although the idea of electrochemical methane upgrade did not prove successful, we have acquired expertise in several key electrochemical and analytic techniques, e.g., cyclic voltammetry and mass spectrometry. The DHA/thermochemical cycle direction could develop into a key thrust in my research group.










