Reports: UR152231-UR1: Boron-Based Directing Groups for Directed Lithiation Reactions
 printer friendly
printer friendly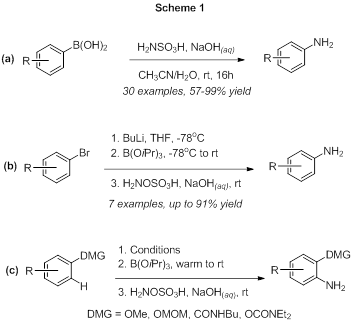
In the area of boronic acid catalysis (BAC), we have developed several new methods based on the activation of alcohols. The use of tetrafluorophenylboronic acid catalyst 1a for Friedel-Crafts (FC) reactions of allylic, propargylic and benzylic alcohols resulted in a substantial improvement in catalytic activity relative to previously reported catalyst 1b (Scheme 2).[2] This effect, which is attributable to the substitution of a single ortho fluorine with hydrogen, occurs despite a significant increase in pKa (pKa(1b)=3.5; pKa(1a)=6.0). Detailed mechanistic studies are ongoing, but preliminary results suggest that the difference in catalytic activity is due to either transition state stabilizing ortho-C-H---O hydrogen bonding with 1a, or destabilizing dipole-dipole interactions between the oxygens in the borate leaving group and the additional C-F bond in 1b.
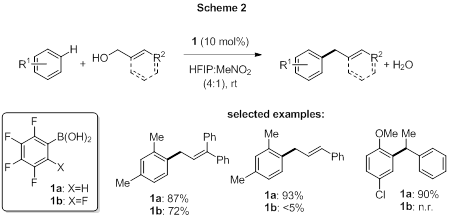
Our recent report on this method features a substantial substrate scope (35 examples), including unactivated arene nucleophiles and those bearing a combination of electron-withdrawing and electron-donating groups. Deactivated benzylic alcohols were also found to be suitable electrophilic substrates for this reaction.
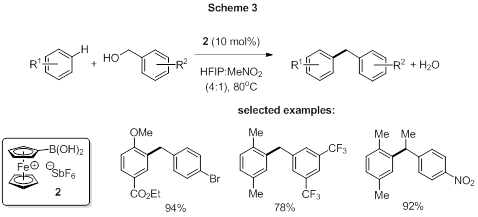
We extended our work on BAC for FC reactions by developing a ferrocenium boronic acid catalyst (2, Scheme 3) for alcohol activation.[3] Not only did we find that 2 is superior to both 1a and 1b, in terms of the scope of both the arene nucleophiles and alcohol electrophiles tolerated, it also out-performs the most effective conventional Lewis acid catalysts for this type of reaction.[4] The efficacy of 2 as a catalyst in this reaction does not appear to be due to enhanced acidity, since its pKa (5.9) is comparable to that of 1a and 1b. Preliminary investigations suggest that ion-pairing effects between the presumed alcohol-derived carbocation intermediate and the SbF6 counter-ion of the catalyst are critical to the observed catalytic activity. Further mechanistic work is ongoing in our lab.
In efforts to expand on the utility of BAC for the activation of alcohols, we are also investigating its application to the recently discovered aza-Piancatelli rearrangement.[5] This reaction, which involves the highly diastereoselective rearrangement of furfuryl alcohols in the presence of anilines, requires the use of a Lewis acid catalyst that is compatible with the Lewis basic aniline substrates. As a result of this requirement, few reports on the use conventional Lewis acids (e.g. Dy(OTf)3, Ca(NTf2)2) have appeared.[6] Even where such catalysts are effective in promoting the reaction, high temperatures are required, and competing adduct formation of the catalyst is rate-limiting.

Screening of reaction conditions and catalysts 1-2 for this reaction revealed that 1a is effective in promoting the aza-Piancatelli rearrangement for a variety of furfuryl alcohols and substituted anilines at ambient temperature (Scheme 4). We have so far demonstrated a moderate substrate scope (20 examples) for this reaction. Preliminary mechanistic investigations suggest that the reaction promoted by BAC proceeds by a different mechanism than that promoted by conventional Lewis acid catalysts and that adduct formation in the former case is not a contributor to the overall rate of reaction. Further synthetic and mechanistic work on this and related reactions are ongoing in our lab.
([1]) Voth, S.; Hollett, J. W.; McCubbin, J. A. J. Org. Chem. 2015, 80, 2545.
([2]) Ricardo, C. L.; Mo, X.; McCubbin, J. A.; Hall, D. G. Chem. Eur. J. 2015, 21, 4128.
([3]) Mo, X.; Yakiwchuk, J.; Dansereau, J.; McCubbin J. A.; Hall, D. G. J. Am. Chem. Soc. 2015, 137, 9694.
([4]) See, for example: (a) Iovel, I.; Mertins, K.; Kischel, J.; Zapf, A.; Beller, M. Angew. Chem. Int. Ed. 2005, 44, 3913.; (b) Rueping, M.; Nachtsheim, B. J.; Ieawsuwan, W. Adv. Synth. Catal. 2006, 348, 1033.
([5]) Veits, G. K.; Wenz, D. R.; de Alaniz, J. R. Angew. Chem. Int. Ed. 2010, 49, 9484.
([6]) Piutti, C.; Quartieri, F. Molecules 2013, 18, 12290.










