Reports: ND151929-ND1: Self-Assembly of Catalysts and Ligands by Reversible Covalent Interactions of Organoboron Compounds
 printer friendly
printer friendlyProgress Report
Introduction. Methods for the rapid synthesis of structurally diverse ligands or catalysts can significantly accelerate the pace of reaction discovery and optimization. A powerful approach involves the self-assembly of candidate structures through noncovalent or reversible covalent interactions such as metal–ligand coordination, hydrogen bonding and ion pairing. Catalysts are generated by simple pairwise mixing of components, thus obviating the need for time-consuming purification and isolation steps. This project explores applications of reversible covalent interactions between boronic acids and diols as the basis for catalyst assembly. Organoboron–diol interactions are specific, strong and reversible, and have been exploited extensively in the molecular recognition field. The tolerance of these reversible covalent interactions to a range of solvents and reaction conditions, and the availability of diverse, chiral diol feedstocks constitute unique potential advantages in the context of applications in catalyst self-assembly.
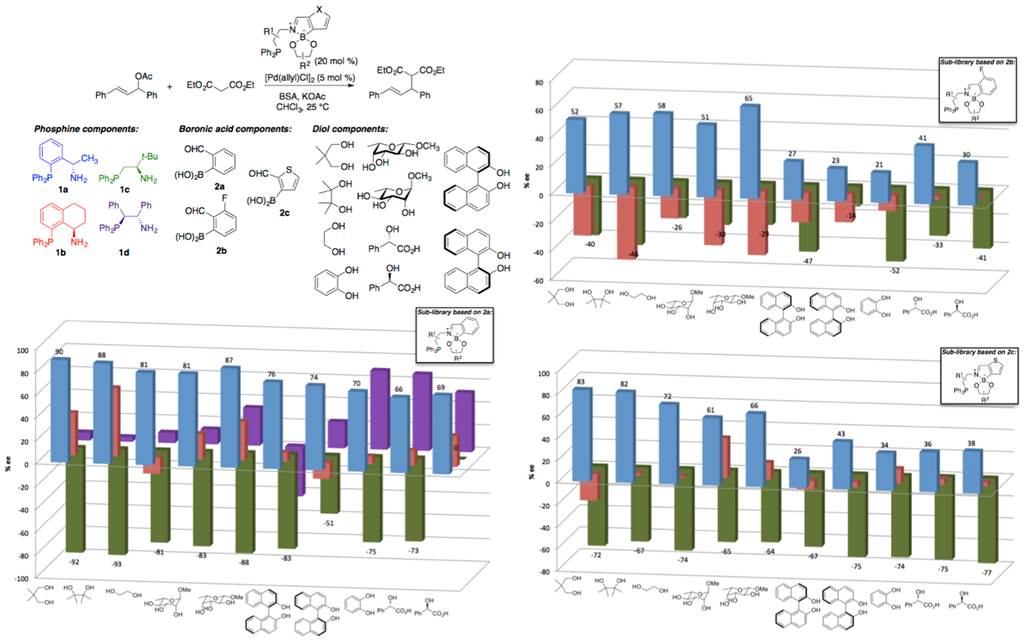
Libraries of chiral phosphines prepared by three-component condensations of 2-formylphenylboronic acid. Our initial efforts focused on the use of boronic acid–diol interactions to guide the assembly of chiral phosphine ligands. However, challenges in accessing the required boronic acid-functionalized phosphine components impeded our ability to generate significant diversity using this approach. We thus adopted a modified strategy, using three-component condensations of phosphine-functionalized amines, diols and 2-formylphenylboronic acid. These reactions generate imine–boronate ligands, with the boronic acid component serving as a linchpin. They proceed rapidly and to very high conversion, generating water as the only byproduct.
Using this approach, we generated a library of 100 phosphines from selected combinations of four aminophosphines, three formyl-functionalized boronic acids and ten diols. The ligands were tested in a Pd(0)-catalyzed allylic substitution reaction (Scheme 1). Each of the three components of the ligand was found to influence enantioselectivity, and matching/mismatching effects were evident for certain pairs of components. A ligand that gave rise to high enantiomeric excess (93% ee) for this benchmark substrate combination was identified.
Scheme 1. Evaluation of a library of 100 self-assembled P,N-ligands for asymmetric allylic alkylation.
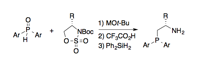
We have investigated applications of this class of self-assembled ligands to other transition metal-catalyzed reactions, and have used a related approach to generate bifunctional organocatalysts that incorporate elements of chirality from sugars, binaphthols, or hydroxy acids. For several of these catalyst types, a chiral b-aminophosphine is a key structural component. Although reliable methods exist for preparing such compounds from enantioenriched b-amino alcohols, variation of the electronic or steric properties of the phosphorus substituents is not straightforward. The ability to make such variations was crucial to our envisioned applications. We thus devised a new method that employs a metalated secondary phosphine oxide as the nucleophile for ring-opening of a cyclic sulfamidate (Scheme 2). The scope of this protocol is broad, and enables the preparation of derivatives having electron-rich, electron-deficient and sterically hindered aryl substituents at phosphorus. We have investigated the effect of these aryl group substitutions on the activity of aminophosphine-based ligands and organocatalysts. More broadly, we anticipate that these novel substituted derivatives will be useful building blocks for other researchers engaged in asymmetric catalysis.
Scheme 2. Access to substituted b-aminophosphines from secondary phosphine oxides.
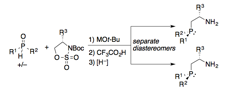
We have exploited the secondary phosphine oxide-based synthetic route detailed above to prepare a new class of diastereo- and enantiomerically enriched, P-chiral b-aminophosphines (Scheme 3). These are synthesized by coupling an unsymmetrical, racemic secondary phosphine oxide with an enantioenriched sulfamidate, generating a mixture of diastereomers that can be resolved by fractional recrystallization or by chromatography. Methods for the synthesis of P-chiral phosphines are relatively rare, and our newly developed protocol is both modular and operationally simple. Our preliminary results indicate that P-chiral b-aminophosphine-based catalysts can provide enhanced stereoselectivities relative to derivatives lacking a P-chiral moiety. Ongoing work is aimed at establishing the generality of this approach and in generating modular and self-assembled P-chiral ligands using these new building blocks.
Scheme 3. P-Chiral b-aminophosphines.