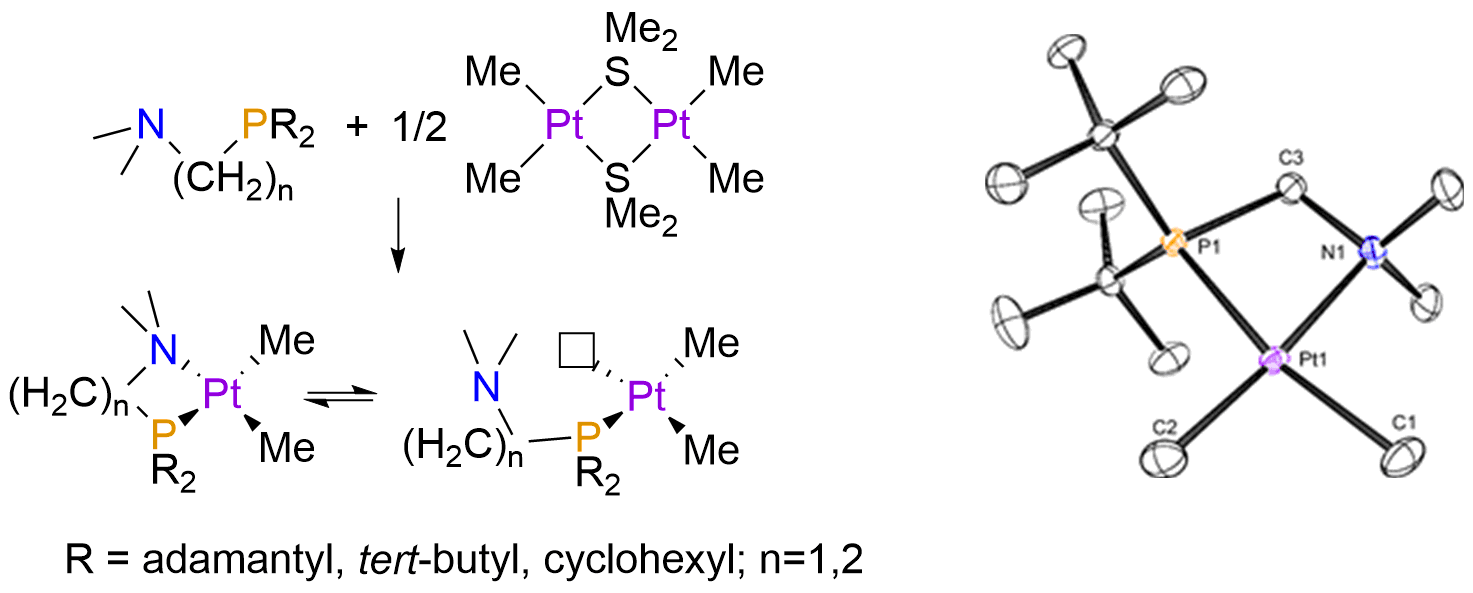Reports: ND353316-ND3: Towards Pt-Catalyzed C-C Bond Formation of Hydrocarbons
 printer friendly
printer friendlyWe have been exploring each of the steps of the proposed Pt-catalyzed conversion of methane into toluene or ethane (depending on the other reactants). We are evaluating the best ligands for each step of the process, with the hope that one ligand will work in each fundamental step of the proposed catalytic cycle.
Reductive elimination from high valent PtIV complexes:
High valent PtIV complexes are critical intermediates in many Pt-mediated transformations, including, but not limited to C-H activation, Negishi-type C-C coupling reactions as well as C-O functionalization. Herein we report our recent investigations of reductive elimination leading to (i) C-C bond formation with unprecedented selectivity, (ii) ligand enabled fast and reversible H/D scrambling in Pt-bound hydrocarbyl groups and (iii) selective B-H to B-O transformations.
(i) Based on earlier reports from the Love group, we investigated a series of coordinatively unsaturated PtIV complexes1 (1-3) bearing both Csp3 and Csp2-hybridized hydrocarbyl fragments, and featuring ligand backbones with varying degrees of flexibility. When subjected to thermolysis in non-coordinating solvents under relatively mild conditions we found that 1 cleanly eliminated ethane. While earlier reports subscribe to the trend (Csp2-Csp2 > Csp2-Csp3 > Csp3-Csp3) of reductive elimination, we anticipated that this unexpected selectivity (Csp3-Csp3) was due to meridional arrangement of hydrocarbyl fragments around PtIV center in 1. To corroborate this result, two other PtIV complexes (2 and 3) featuring greater flexibility of the ligand scaffold were investigated. We found in both of these cases that the original trend of reductive elimination could be reinstated. Through a comparative experimental and theoretical study, we show that the rigidity a meridionally coordinating ligand can change the intimate mechanism of reductive elimination. A manuscript will be submitted to JACS in early October that details these results.
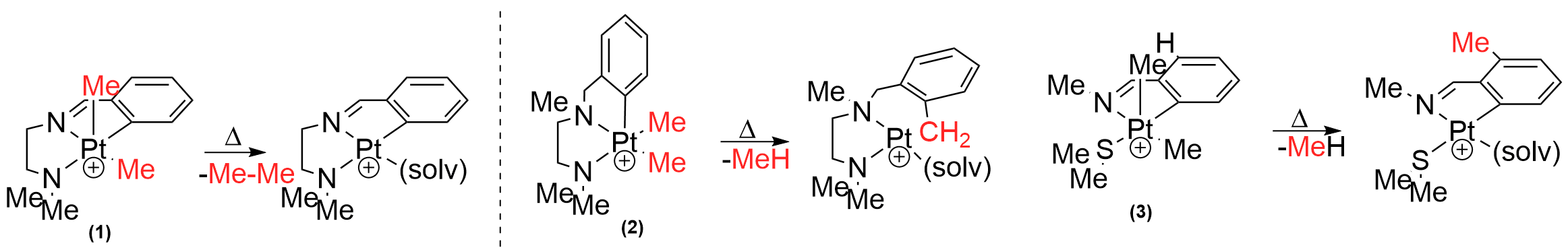
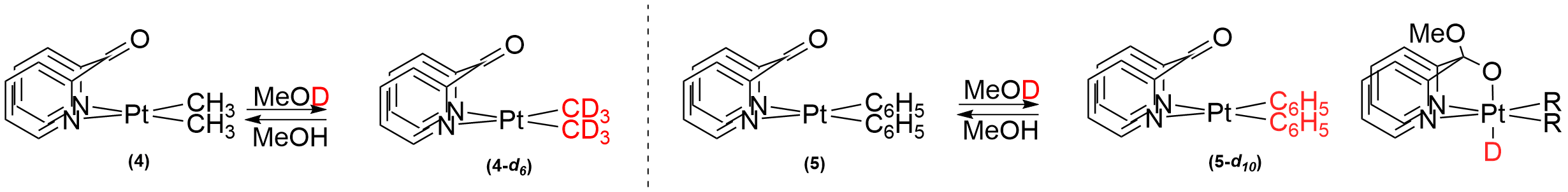
(ii) We demonstrated facile H/D exchange between CD3OD and Pt-bound hydrocarbyl fragments in 4 and 5. Although similar reactivity with protic solvents has been reported, to the best of our knowledge, all previously documented systems have finally undergone irreversible loss of hydrocarbon (methane or benzene). Owing to no loss of hydrocarbon, the reaction could be reversed: dissolution of the perdeutero complexes (4-d6 or 5-d10) in CD3OH led to full recovery of the starting materials. We have performed extensive kinetic investigations and support our proposed mechanism involving a reversibly formed transient PtIV hydride through DFT calculations. A manuscript will be submitted to Eur. J. Inorg. Chem. (invited) in mid October that details these results.
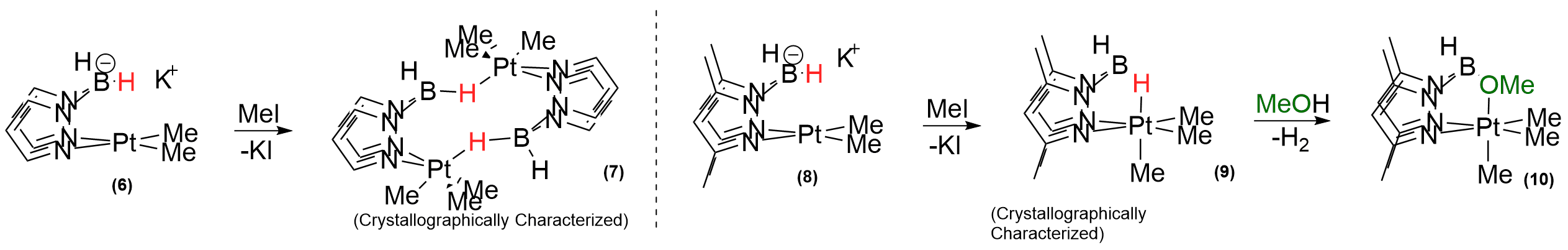
(iii) In continuation of our efforts to access PtIV hydrides, we synthesized a series of PtII complexes supported by well known Bis(pyrazolyl)borohydride ligands of the heteroscorpionate class. Earlier reports of supported Pt complexes is however limited. Oxidation of the PtII center in 6 with methyl iodide led to the formation of a novel intramolecular agostic B-H-Pt interactions in a binuclear complex (7), which was crystallographically characterized. Interestingly, adding steric bulk to the ligand backbone via 3,5 methyl substitutions as in 8 led to the formation of a mononuclear analogue, 9. The bridging hydride in 9 was found to be quite hydridic: fast protonolysis in methanol yields the methoxy complex 10. As a testament to the enhancement of hydridicity of the BH fragment upon coordination to the PtIV center, no protonolysis of the non-interacting (exo) BH fragment in 9 was observed in the course of the reaction. A manuscript will be submitted to JACS in by the end of 2015 that details these results.
Synthesis of mixed-donor P,N ligands for hydrocarbon functionalization:
Both experimental{Lersch, 2005 #1180} and theoretical works have established that platinum(II) complexes bearing amine ligands are ideal for C-H activation, but don’t promote C-C reductive elimination at low temperatures. On the other hand, bidentate phosphine complexes allow for facile C-C bond formation, but are not as effective at cleaving C-H bonds. We have been working toward the development of platinum(II) complexes bearing mixed-donor P,N complexes that should impart intermediate properties to the metal center (Figure 1b), allowing for catalysis. By modulating the sterics and bite angle of the P,N ligands, we have prepared complexes that are hemilabile, with reversible dissociation of the nitrogen arm allowing for reactivity with small molecules. As desired, we found that reductive elimination of C-C bonds from platinum(IV) complexes of these ligands was facile at room temperature, readily forming ethane from (P,N)PtMe3I. Reactivity studies with CO and isonitriles show that small molecules readily displace the nitrogen arm of the P,N ligand, a promising result that suggests that hydrocarbon activation may even be possible without the removal of one of the anionic hydrocarbyl groups. The hemilability of the nitrogen arm in these complexes may also allow for catalytic hydroarylation or hydroalkylation olefins (via substitution for an olefin at the amine position), an area that we are also actively investigating. A manuscript will be submitted to JACS in early 2016 that details these results.