Reports: DNI352704-DNI3: Designing Multifunctional Complexes for Chemical Catalysis
 printer friendly
printer friendlyThe long term goal of this project is to delineate the parameters that affect the cooperative reactivity of metal ions within clusters. Understanding how to design and control redox cooperativity within clusters will lead to new catalytic systems for challenging multielectron redox reactions, such as dinitrogen reduction and O2 activation, and these metal cluster catalysts will likely require less energy to execute these reactions than conventional methods. These basic redox reactions are pivotal for renewable fuel synthesis (e.g., CO2 reduction) and hydrocarbon functionalization (e.g., methane oxidation to methanol), among other applications. Our approach is based on the design of modular macrobicyclic ligands to template cluster assembly; therefore, using a simple rational design approach we can systematically investigate how electronic and steric effects dictate reactivity. Our goal for this proposal was to develop the chemistry of macrobicyclic ligands and to demonstrate the efficacy of our approach.
Prior to the reporting period, we communicated the synthesis of a tris(diketimine) cyclophane ligand (H3L) and a hexacarboxamide cryptand (H6L’), which both template trinuclear clusters. This designed approach to clusters using macrobicycles was demonstrated in the synthesis of triiron and tricopper cryptate complexes, the selective synthesis of dimetallic cryptate complexes with proximal hydrogen bonding interactions, and the synthesis of the triiron(II) tri(bromide), trimanganese(II) tri(bromide), trichromium(II), tricobalt(II), trinickel(II), tricopper(I), trizinc(II), trigermanium(II), tritin(II), and trialuminum(III) cyclophane complexes. In addition, we communicated our early findings on dinitrogen reduction upon reduction of our triiron complex and coordination of dinitrogen by the tricopper(I) complex (Figure 1). These results are the first examples of a designed iron cluster capable of N2 activation and of a complex containing Cu–N2 interactions; these results were published after the reporting period. We also reported preliminary reaction surveys of a reduced chalcogenide-bridged tricopper cluster with nitrous oxide and dioxygen, and that of the tricopper(I)-dinitrogen complex with azide and nitrous oxide. These reactivity studies are still ongoing in our research program.
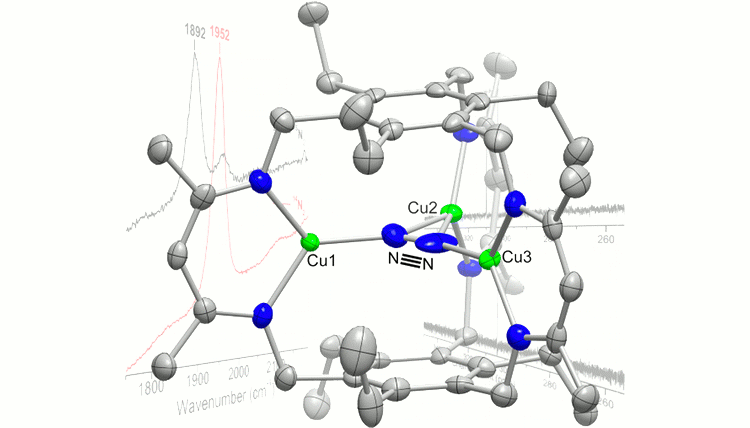
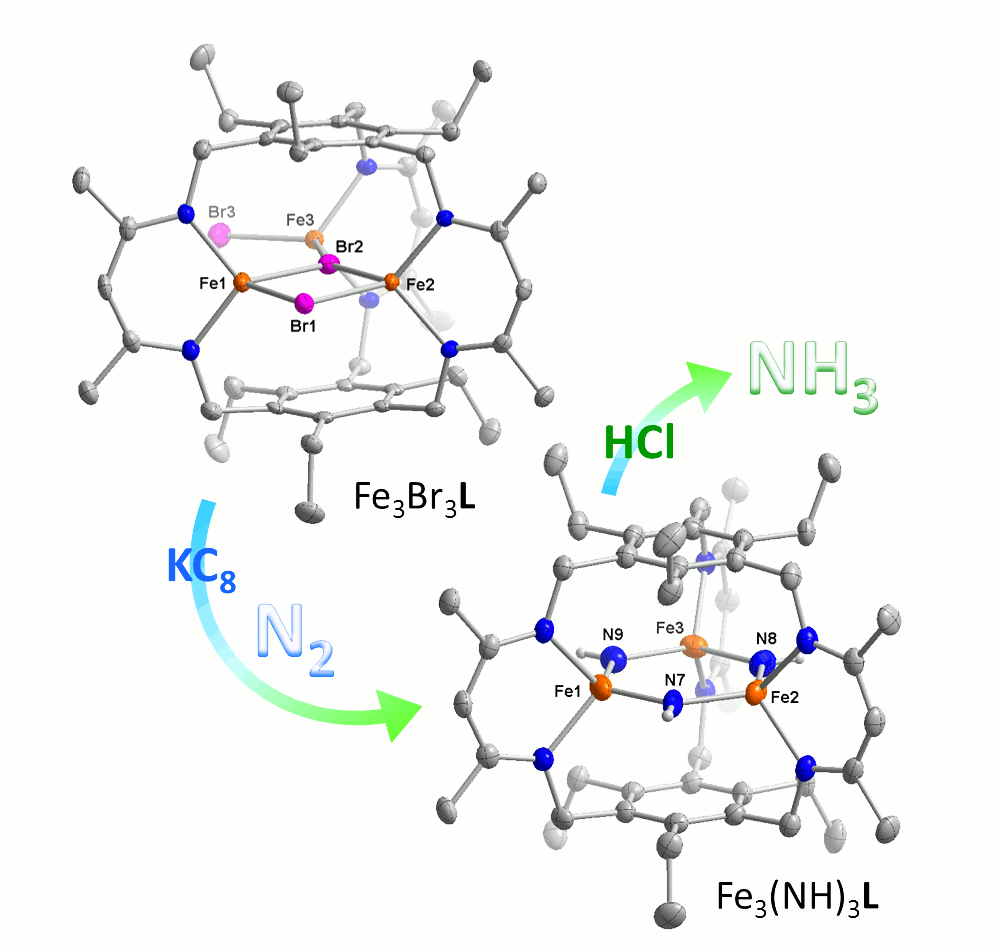
Figure 1. Tricopper(I)-dinitrogen complex (left) and reaction scheme and products from reduction of a triiron(II) cluster in the presence of N2.
We previously reported that the reaction of Fe3Br3L with KEt3BH or Ph3CS- affords the trihydride complex, Fe3H3L, or the triferric tri(sulfide), Fe3S3L, clusters. In addition, we indicated that Fe3H3L reacts specifically with CO2 to give Fe3H2(OOCH)L (Figure 2), but the trihydride is unreactive towards all other substrates tested (inc., CH3CN, CS2, C2H2) at room temperature. We have since demonstrated and published that the tricobalt trihydride analog also exhibits similar specificity; however, whereas heating the Fe3H3L in the presence of CO2 results in all hydrides reacting to give Fe3 (OOCH)3L, we only isolate the monoformate product for the tricobalt system under all conditions tested. We then probed the reaction of the trizinc trihydride analog (Figure 2), and reported the first example of an air-stable and water tolerant ZnH complex. Moreover, this compound showed unprecedented specificity for CO2, much like the iron and cobalt congeners. With respect to iron-sulfur cluster chemistry, we also added a new member to these class of compounds: a (m2-bromo)(m3-sulfido)triiron(II) cluster. This species represents a unique example of a coordinatively unsaturated iron-sulfur cluster, and structurally bears similarities to the reactive iron centers in the iron-molybdenum cofactor in Mo-dependent nitrogenases.
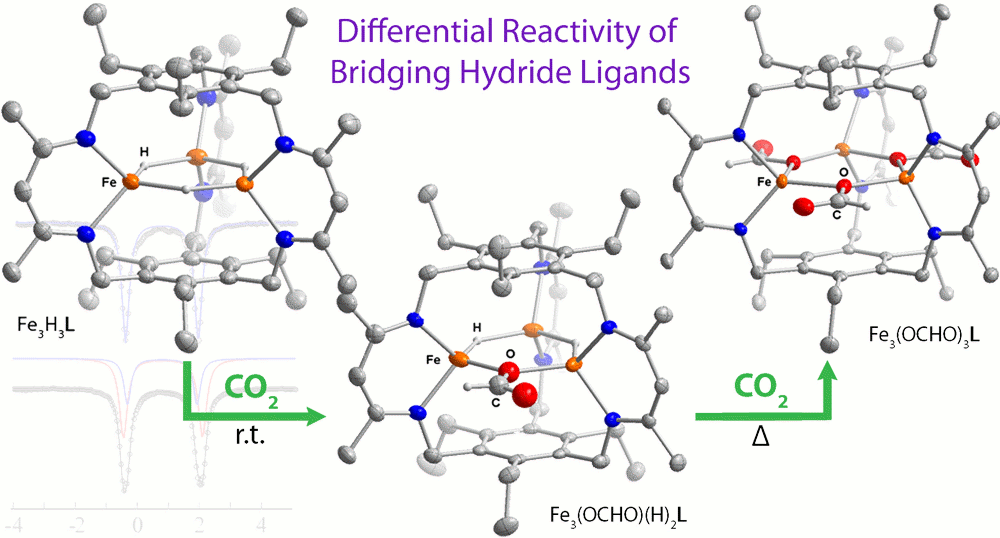
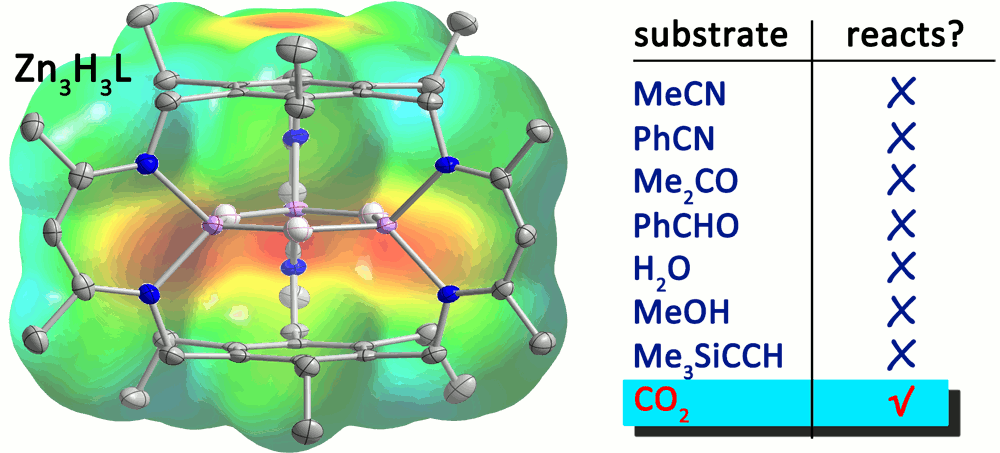
Figure 2. Reaction of the triiron(II) trihydride complex with CO2 (left) and the trizinc(II) trihydride complex with its corresponding substrate table.
Finally, we discovered that the tricopper(I)-dinitrogen cluster reacts rapidly with O2 to generate oxygenated intermediates that are capable of O-atom insertion into benzylic C–H bonds, and transfer an O-atom to styrene and thioethers. The efficiency of O-atom transfer is limited, likely by competitive ligand decomposition, but nonetheless represents a rare example of a synthetic copper cluster that effects O-atom transfer to exogenous substrates; typically, H-atom abstraction is observed. This type of reactivity resembles the mechanism of methane oxidation by the dicopper cluster in particulate methane monooxygenase, and suggests that synthetic copper complexes can be designed with sufficient oxidizing power to activate C–H bonds in hydrocarbon substrates.










