Reports: ND1054075-ND10: Designing Lithium Based Hard Materials from First Principles
 printer friendly
printer friendlyNarrative Progress Report
The main goal of this project is to investigate novel and stable phases of lithium alloys that can be of use in energy applications, hard, and light structural materials. For such purposes, we use first principle calculations and advance structural search methods to identify stable structural configurations.
Li-Si alloys
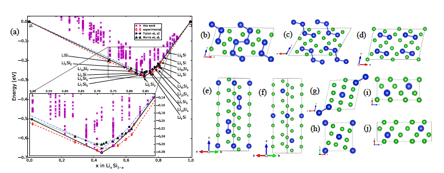
Figure 1 (a) Convex-hull of the lithium-silicon binary system. The black continuous line is obtained from ab-initio MHM calculations where the black circles indicate thermodynamically stables structures. The red dashed line is the convex-hull constructed by using structural experimental data to obtain the formation energy (red diamonds). The blue dashed-dotted line and green dotted line are the theoretical convex-hulls (blue star and green triangle symbols) reported by Morris et al. and Tipton et al., respectively. Crystal structure of (b) Li3Si2 (monoclinic, C12m/1), (c) Li2Si (monoclinic, C12m/1), (d) Li9Si4 (monoclinic, C12m/1), (e) Li5Si2 (trigonal, R-3m) (f) Li6Si (trigonal, R-3m) (g) Li3Si (monoclinic, P12/m1), (h) Li4Si (tetragonal, I4/m), (i) Li7Si2 (trigonal, P-3m1) and (j) Li5Si (trigonal, P-3m1). Li atoms are green while Si atoms are blue.
For Li-Si system we have studied sixty different stoichiometries with unit cells containing up to 16 atoms, starting from pure silicon and changing the stoichiometry up to pure lithium. Combining the calculated formation energies for all different chemical compositions, we have calculated the theoretical convex-hull as depicted in Fig. 1a. This plot summarizes the most chemically stable stoichiometries. Direct comparison with other theoretical results as well as with experiments has also been performed. The agreement with previous reports and known experimental data is quite good. By inspecting Fig 1a, we see that our theoretical convex-hull lies above the experimental structures for Li12Si7, Li7Si3, Li13Si4, Li22Si5, Li21Si5 and Li15Si4. This is basically due to the fact that the experimental crystal unit cells contain more than 16 atoms. Additionally to the reported structures, we have also found a large set of structures, which has not been reported previously. In fact, our calculations have identified two new phases: one for Li3Si (space group P12/m1) and other for Li4Si (space group I4/m), which belong to the convex hull. Additionally, we found novel low-energy meta-stable structures for Li3Si2 (space group C12/m1), Li2Si (space group C12/m1), Li9Si4 (space group C12/m1), Li7Si2 (space group P-3m1), Li5Si (space group P-3m1) and Li6Si (space group R-3m), which are very close to the convex-hull (only 4, 3, 5, 2 and 4 meV/atom, respectively). These energy differences are on the order of the numerical accuracy of our theoretical description. Moreover, meta-stable structures close to the convex-hull could be easily stabilized experimentally by temperature, pressure, or doping. The atomic arrangement that we obtained for Li3Si2, Li2Si, Li9Si4, Li5Si2, Li3Si, Li7Si2, Li4Si, Li5Si and Li6Si are further displayed in Fig. 1b-j. For all the structures reported here, we observe that all phonon frequencies are positive, whence unstable modes are not present, which means that these structures are dynamically stables.
Li-Au alloys
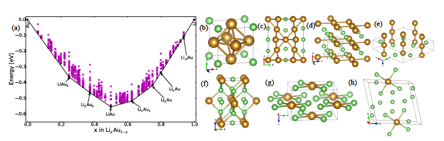
Figure 2: (a) Convex-hull of the Lithium-Gold binary system obtained from MHM calculations. The black circles indicate thermodynamically stables structures. Crystal structures for (b) LiAu2 monoclinic, P121/m1, (c) LiAu Orthorhombic, Pmmm, (d) Li5Au3 hexagonal, P-62m, (e) Li3Au tetragonal, I4/mmm, (f) Li4Au tetragonal, I4/m (h) Li15Au monoclinic, P-1. Li atoms are green while Au atoms are yellowish.
As in the case of Li-Au, we have also used unit cell not larger than 16 atoms. The theoretically calculated convex-hull is depicted in Fig2a. The crosses represent structures with lowest energy while the circles represent predicted thermodynamically stable phases. First of all, we were able to obtain the experimentally reported structures for LiAu3 (Pm-3m), LiAu (Pm-3m) and Li3Au (Fm-3m). Additionally, we found new crystal structures with symmetries C12/m, P-26m, I4/m and P-2 for Li3Au5, Li5Au3, Li4Au and Li15Au respectively. These new configurations have not been reported previously. In the particular case of LiAu and Li3Au, we found structures with lower energy structures than previously reported (although with differences close to 5 meV), with space groups P/mmm and I4/mmm respectively. The arrangement of atoms for optimal structures is shown in Fig2b-h. For all these structures we calculate the phonon dispersion, we observe that all these structures are dynamically stable. This convex-hull will also inform us about possible crystal transformations as the lithium content is changed in the system, a process performed in the lithium batteries.
Bi-Sb alloys
In recent decades, Bi1_xSbx alloys have attracted considerable interest due to their unique electronic properties. These alloys are among the best-known thermoelectrics at low temperatures and are considered within the first generation of topological insulators. Being an alloy, Bi1_xSbx has random substitutional disorder, which makes its electronic structure and surface-states very complex. Due to such complexity of the electronic structure, it is very difficult to describe the electronic properties of Bi1_xSbx alloys using simple theoretical models. Since the electronic properties of materials mainly depend on their crystal structure, it is very important for us to find the stoichiometric crystals of Bi1_xSbx with ordered atomic configurations. Therefore, this problem is ideal for the methods used in this project.
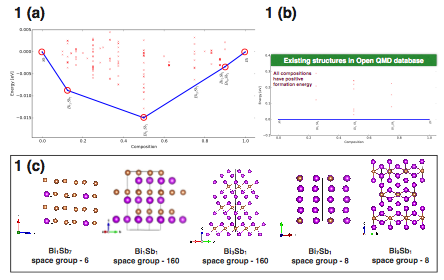
Figure 3: (a) Calculated Convex-Hull for the BixSb(1-x) alloy. Red circles denote the stable configurations. (b) Convex-hull reported on the QMD database based on an exhaustive search. Notice that there is not a single predicted stable structure. (c) Crystal structures of the found stable configurations.
Figure 3(a) represents the calculated convex-hull for Bi1-xSbx. Four different stoichiometries are on the convex-hull, all with negative formation energy. The Bi1Sb1 composition (space group 160) has the lowest formation energy; hence it would be the most stable structure for Bi1-xSbx alloy. Interestingly, we notice that by applying pressure on Bi1Sb1 compound, one can realize a Weyl semi-metallic phase in this system. Such realization of Weyl semi-metallic phase in this non-magnetic system is very exciting, which is of large interest in the science community. While the crystal structure for the one/one composition was already known, the other stoichiometries are new and posses interesting electronic properties. Some of the obtained low energy crystal structures are shown in figure 3(c).










