Reports: ND152737-ND1: Catalytic Asymmetric Carbon-Carbon Bond Formation with Fluoroenolates Generated Detylated Prenucleophiles
 printer friendly
printer friendlyThe
use and need of chiral organofluorine compounds in
medical applications has dramatically increased in recent years. The asymmetric
production of fluorinated fine chemicals, however, is quite challenging and new
catalytic methodologies that utilize fluorinated intermediates are in high
demand. General rules and methods developed for asymmetric transformations with
nonfluorinated substrates and reagents can often not
be applied to structural analogues carrying a monofluoromethylene
unit or an even more dominant difluoromethylene or trifluoromethyl group next to the reaction center. The
strong stereoelectronic effects exerted by proximate
carbon-fluorine bonds typically impede established synthetic protocols and may
even alter reaction pathways. A specific goal of this proposal is therefore to
introduce new ways to generate reactive fluorinated enolates
from readily available starting materials and to employ these in catalytic
asymmetric C-C bond formation. Inspired by Colby’s work,
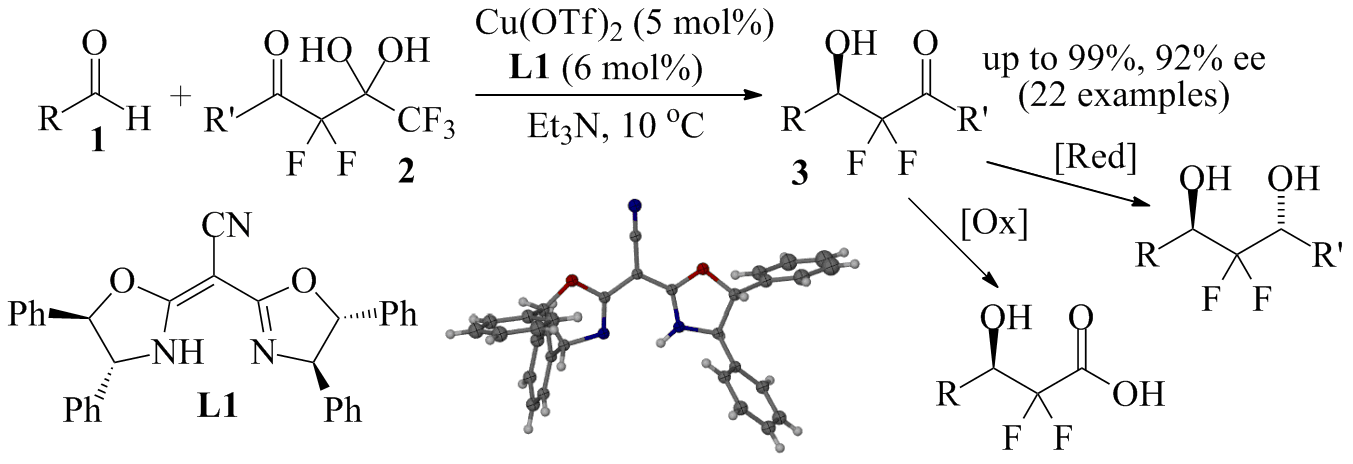
Scheme 1. Catalytic enantioselective difluoroalkylation of aldehydes.
Since then, we have extended this approach to 4 and other fluorochloro analogues of 2. Despite extensive screening of solvent, base, catalyst and temperature, we have not found conditions that produce the corresponding aldol reaction products 5 with high stereoselectivity (Scheme 2). This is probably due to a lack of control of the E/Z isomerism of the intermediate chlorofluoroenolate. However, we have been able to trap the enolate with N-bromosuccinimide, NBS, to prepare a small series of the trihalogenated bromochlorofluoromethyl ketones 6 in very good yields (Scheme 3). While these compounds are highly susceptible to haloform reactions and not stable for a long time at room temperature, we have found that they undergo Horner-Wadsworth-Emmons (HWE) and Corey-Fuchs reactions to the corresponding alkenes. The HWE reaction between 6a and phosphonate 7 gave the a,b-unsaturated ester 8 in 90% yield and with excellent diastereoselectivity. The determination of the cis/trans configuration of 8 is underway. Similarly, the Corey-Fuchs transformation occurs under mild conditions without noticeable side reactions and we obtained the dibromoalkene 9 in 98% yield. We are currently investigating the full scope of this chemistry and we are planning to explore the possibility of catalytic asymmetric bromination of the chlorofluoroenolate prepared in situ from the diol 4. Finally, the use of racemic or enantioenriched (if available) 6, 8 or 9 in palladium catalyzed cross-coupling reactions will be studied. We envision that this could provide unprecedented access to a variety of chiral compounds carrying a central chlorofluoromethylene unit.
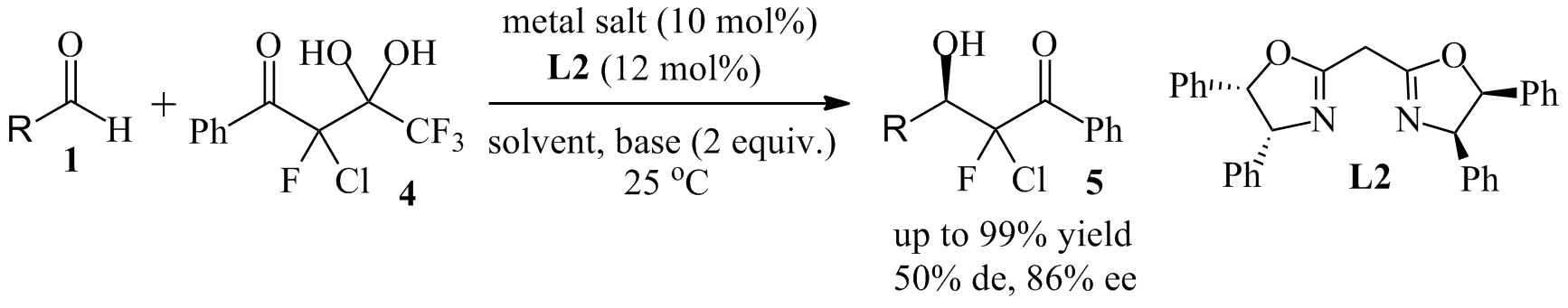
Scheme 2. Catalytic aldol reaction with chlorofluoro diol 4.
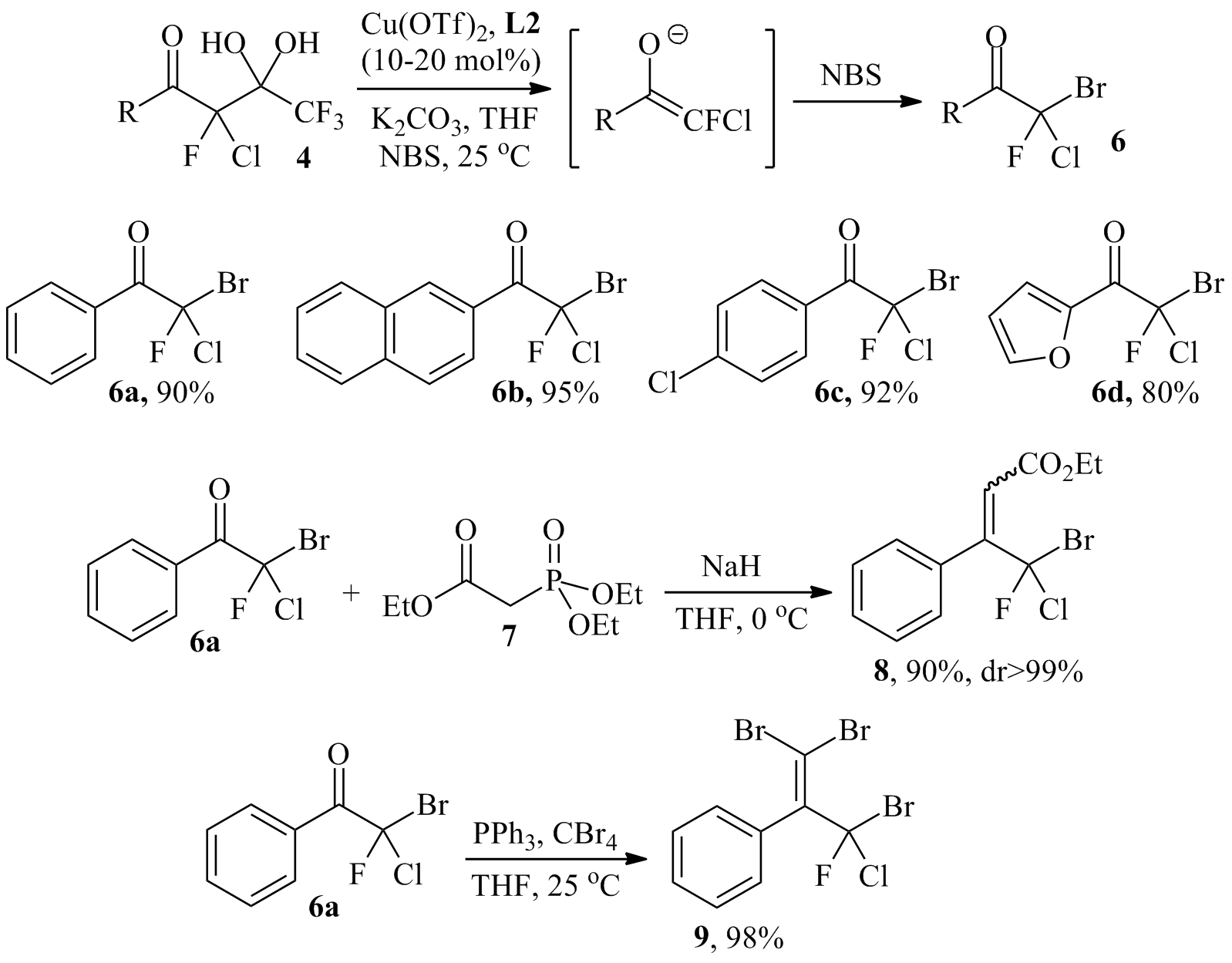
Scheme 3. Top: Conversion of chlorofluoroenolates generated in situ via base-promoted C-C bond cleavage of 4 and structures of the products formed with NBS. Bottom: Horner-Wadsworth-Emmons synthesis of 8 and Corey-Fuchs reaction forming 9.
We have encountered an efficient asymmetric allylic alkylation method that utilizes fluorooxindoles 10 which have been used as valuable building blocks to make biologically active heterocyclic compounds. Enantioselective synthesis of 3-fluorooxindoles has been achieved mostly by late stage fluorination of activated oxindoles. However, many of these procedures are limited to few substrates. In contrast, the use of 3-fluorooxindole as nucleophile in enantioselective transformations is less explored. Through screening of a wide range of transition metal catalysts, chiral ligands, temperature effects, base and solvents, we have developed a highly enantioselective synthesis of 3-fluoro-3-allyloxindoles 12 that is based on palladium mediated allylic alkylation with the readily available allylic acetate 11. The reaction occurs smoothly at room temperature with quantitative conversion and shows no sign of by-product formation. We note that the product contains a quaternary chiral center exhibiting a fluoride, and it has several synthetically useful functional groups that may prove invaluable for further derivatization. At this point, the yield and the enantiomeric excess are generally above 90%. We are in the final stage with the optimization of the reaction conditions to improve the diastereoselectivity. We will then determine the scope and general utility of this reaction.
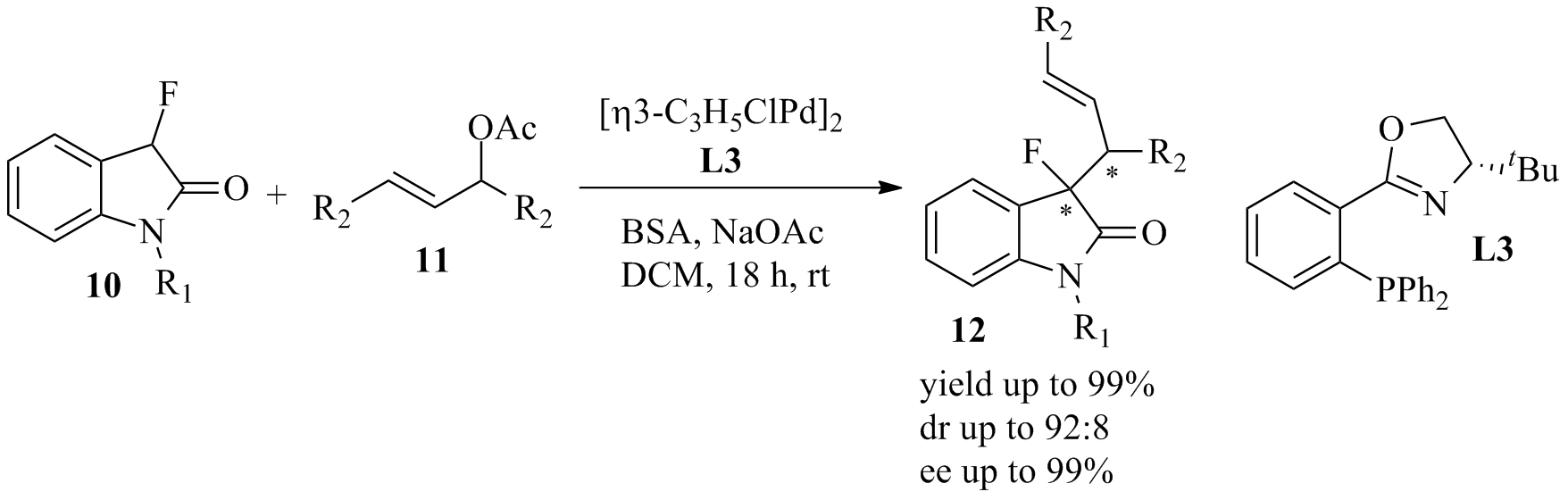
Scheme 4. Asymmetric allylation of fluorooxindoles.
Our methodical approach to
asymmetric catalysis with fluoroenolate intermediates
formed via base-promoted cleavage of diols such as 4 has received considerable attention and been applied in multiple
ways by several research groups. For example, Pan et al. used essentially our
method in a related aldol reaction producing fluorinated products with two
adjacent chiral centers.
[1] Han, C.; Kim, E. H.; Colby, D. A. J. Am. Chem. Soc. 2011, 133, 5802-5805.
[2] Zhang, P.; Wolf, C. Angew. Chem. Int. Ed. 2013, 52, 7869-7873.
[3] Xie, C.; Wu, L.; Han, J.; Soloshonok, V. A.; Pan, Y. Angew. Chem. Int. Ed. 2015, 54, 6019-6023.
[4] Zhang, P, Liu, Y.; Wolf, C. Asymmetric catalysis with fluorinated substrates and in situ generated haloenolates. ISCD, Boston, MA, USA, 2015.










