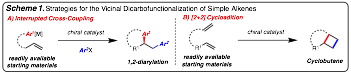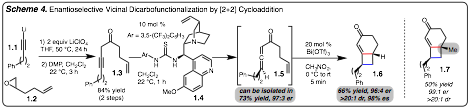Reports: DNI154422-DNI1: Cu-Catalyzed Vicinal Dicarbofunctionalization of Simple Alkenes
M. Kevin Brown, Ph.D., Indiana University
Overview: This PRF grant has been used to support two distinct, but related,
projects within the group: 1) Cu-catalyzed interrupted cross-coupling reactions
(Scheem 1A), and 2) catalytic enantioselective [2+2]
cycloadditions of unactivated alkenes (Scheme 1B). Both of these methods serve to provide
solutions to the contemporary challenge of functionalization of the
petrochemical resource, unactivated alkenes. This was a key objective that was
outlined in the PRF proposal.
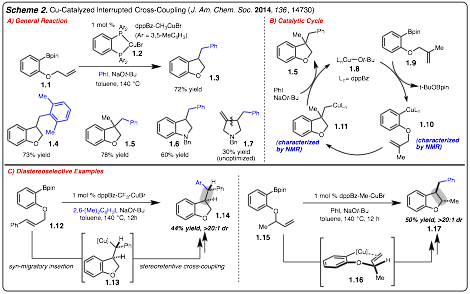
1) Cu-Catalyzed Interrupted Cross-Coupling: Over the last year we have made significant advances toward
development of Cu-catalyzed diarylation of alkenes by
interrupted cross-coupling (i.e., vicinal dicarbofunctionalization of alkenes). We have identified that treatment of readily
prepared substrate 1.1 with
Cu-catalyst 1.2, PhI
and NaOt-Bu
led to the formation of benzofuran in good yield
(Scheme 2A). This process has been
extended to various aryl iodides (e.g., 1.4)
and to the formation of nitrogen heterocycles (e.g., 1.6-1.7). The catalytic
cycle of this reaction has been determined by isolation of key intermediates
and proceed by the following elementary steps: 1) transmetallation
to generate Cu-complex 1.10, 2) migratory
insertion of the pendant alkene into the Cu-Ar bond
to furnish 1.11, and 3) capture of
the Csp3-Cu-complex 1.11
with PhI (Scheme 2B).
This method has been extended in two key
directions: 1) diastereoselective
variants and 2) enantioselective transformations.
With respect to the former, two key observation have been made (Scheme 2C). 1) 1,2-disubstituted alkenes undergo
reaction with high levels of diastereoselectivity (1.12 to 1.14). These results suggest that the process
proceeds by syn-migratory insertion to generation Csp3-Cu-complex
1.13 followed by stereoretentivecross-coupling. 2) Reaction with substrate 1.15 underwent highly diastereoselective diarylation
to generate 1.17, likely proceeding
through pretransition state 1.16 (Scheme 2C).We
have recently obtained preliminary results for catalytic enantioselective
alkene interrupted cross-coupling (Scheme 3). Through the evaluation of a set of
chiral ligands related to dppBz, use of 3 mol % BenzP* 1.22 was effective for converting 1.18 to 1.20 in 55%
yield and 97:3 er (Scheme 3). Furthermore, highly
enantioselective synthesis of quaternary carbons is also possible (e.g., 1.21). A preliminary substrate scope has been
established and found to include a variety of aryl iodides and tolerate the
formation of indoline products (1.24 – 1.26). The future direction of this project aims
to extend the scope of our enantioselective reaction as well as develop fully
intermolecular variants.
2) Enantioselective [2+2] Cyclcoaddition: The studies outlined above describe the vicinal diacarbofunctionalization of alkenes by interrupted cross-coupling. Along these lines our group has also
recently taken an interest in [2+2] cycloadditions as these reaction accomplish
a similar goal, yet also generate a ring.
In order to engage unactivated alkenes in
[2+2] cycloadditions, the unique reactivity of allenes or ketenes needs to be
accessed. This is due to the
orthogonal ¹-bonds that allow for a concerted yet asynchronous [¹2S+(¹2S+¹2S)]
cycloaddition. This initiative has
led to the development of a unique chiral transfer [2+2] cycloaddition of
highly enantiomerically enriched allenic ketones
(Scheme 4).
The overall process is outlined in Scheme
4. An alkynyl
ketone is readily prepared in two steps from a simple epoxide and alkyne. Ketone 1.3 is then treated with thiourea
catalyst 1.4 for 1h to promote an
enantioselective isomerization to provide chiral allene 1.3. This intermediate
can be isolated if desired, or Bi(OTf)3can be added to promote a [2+2] cycloaddition reaction. Overall this method provides access to
substituted cyclobutanes (e.g., 1.6 and
1.7) with high levels of
enantioselectivity. This method
also represents rare example of a chiral transfer [2+2] cycloaddition. Future efforts aim to extend this strategy
to the formation of other ring systems and intermolecular variants.
Conclusions:
Through development of the
two processes outlined above, solutions to the long-standing challenge of vicinal
dicarbofunctionalization of unactivated alkenes have
been provided. A graduate student
will continue to work on the Cu-catalyzed interrupted cross-coupling
with support from the ACS-PRF.
 printer friendly
printer friendly