Reports: ND151929-ND1: Self-Assembly of Catalysts and Ligands by Reversible Covalent Interactions of Organoboron Compounds
 printer friendly
printer friendlyProgress Report
Introduction. Methods for the rapid synthesis of structurally diverse ligands or catalysts can significantly accelerate the pace of reaction discovery and optimization. A powerful approach involves the self-assembly of candidate structures through noncovalent or reversible covalent interactions such as metal–ligand coordination, hydrogen bonding and ion pairing. Catalysts are generated by simple pairwise mixing of components, thus obviating the need for time-consuming purification and isolation steps. This project explores applications of reversible covalent interactions between boronic acids and diols as the basis for catalyst assembly. Organoboron–diol interactions are specific, strong and reversible, and have been exploited extensively in the molecular recognition field. The tolerance of these reversible covalent interactions to a range of solvents and reaction conditions, and the availability of diverse, chiral diol feedstocks constitute unique potential advantages in the context of applications in catalyst self-assembly.
Initial results. After preliminary investigations in which we used boronic acid–diol interactions to guide the assembly of chiral phosphine ligands, we turned our attention to a modified strategy based on three-component condensations of phosphine-functionalized amines, diols and formyl-functionalized boronic acids (Scheme 1). The use of the boronic acid component as a linchpin obviated the need for diverse, boronic acid-functionalized components, which were challenging to synthesize. The three-component nature of this condensation was another advantage for the preparation of libraries of prospective ligands. Using this approach, we generated a library of 100 novel chiral phosphines that we screened as ligands in Pd(0)-catalyzed allylic substitution reactions. Significant variation in activity and enantioselectivity was observed, and certain new ligand architectures were found to give higher than 90% enantiomeric excess for the test reaction (e.g., compound 1).
Scheme 1. Libraries of chiral phosphines generated by three-component condensations of formyl-functionalized boronic acids.
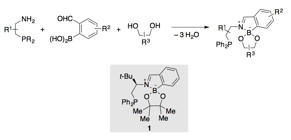
Recent progress. Chiral b-aminophosphines were important building blocks in the three-component ligand assembly strategy described in the preceding paragraph. Because these compounds can be prepared from readily available amino alcohols, several variants are commercially available and others can be synthesized according to published protocols. However, variants having substituents other than phenyl groups at phosphorus are generally not available. Given that variation of steric and electronic properties is a key aspect of phosphine ligand optimization, we set out to develop methods to prepare b-aminophosphine derivatives having a variety of P-aryl substituents. We were able to accomplish this goal by using diarylphosphinites as nucleophiles in ring-opening reactions of amino alcohol-derived sulfamidates (Scheme 2). The key advantage of this method is the ability to purify the relatively stable b-aminophosphine oxide products of this reaction, and then to reduce them cleanly to the corresponding phosphines. Using this method, we were able to synthesize previously unknown b-aminophosphines having electron-donating, electron-withdrawing and sterically hindered P-aryl substituents. We have employed these compounds to explore structure–activity relationships for enantioselective Morita–Baylis–Hillman reactions catalyzed by chiral, bifunctional phosphine–thioureas, and as ligands for transition metal-catalyzed reactions.
Scheme 2. Preparation of substituted b-aminophosphines using diarylphosphinites as nucleophiles.
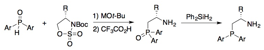
Recently, we have extended this approach to include ring-opening reactions using unsymmetrical diarylphosphinites, leading to diastereo- and enantioenriched P-stereogenic b-aminophosphines (Scheme 3). The diarylphosphinite/sulfamidate coupling generated a mixture of b-aminophosphine oxide diastereomers that could be separated by fractional recrystallization. A novel, amine-assisted reduction enabled stereospecific reduction of the resulting phosphine oxides to the corresponding b-aminophosphines.
Scheme 3. Preparation of diastereo- and enantiomerically enriched, P-stereogenic b-aminophosphines.
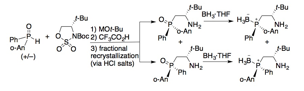
We employed the products of this methodology to synthesize the first examples of bifunctional organocatalysts having a P-stereogenic phosphine moiety (Figure 1). Employing these phosphine–thioureas as catalysts in enantioselective Morita–Baylis–Hillman reactions revealed that the configuration of the P-chirality centre has a marked effect on the activity (but not the enantioselectivity) of the organocatalyst. Given the wealth of transformations that are promoted by phosphine-based bifunctional catalysts, numerous further applications of these P-stereogenic variants can be envisioned.
Figure 1. Organocatalysts based on P-stereogenic b-aminophosphines.
![]()










