Reports: ND153964-ND1: The Development of a New Class of Atropisomeric Heteroaromatic Biaryl Ligands for Enantioselective Transformations
 printer friendly
printer friendlyProgress Report Period: September 1, 2015- August 31, 2016
The proposal entitled described two specific aims:
1. To prepare chiral ligands and complexes with pi-stacking components and probe structural effects. The goal of this aim is to prepare and study chiral ligands by incorporating chelating heteroatoms (P,N,O) into the basic scaffold shown above.
2. To employ chiral complexes with the stabilized biaryl motif in enantioselective reactions. The goal of this aim is to capitalize on the unique properties of heterocyclic, axially chiral biaryls for use as catalysts.
Progress towards Aim 1:
During the last budget period, we were able to establish a reliable and reproducible protocol for the synthesis of our ligand, now dubbed StackPhos (Scheme 1).
Scheme 1
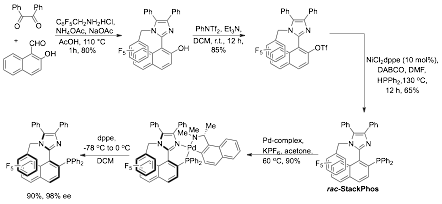
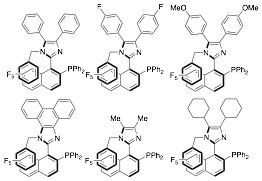
During this reporting period, we utilized this protocol for the synthesis of new ligand congeners (Figure 1) and observed configurational stability of all of these ligands. As shown above, the procedure proved to be amenable to derivative synthesis with minimal changes needed. It is noteworthy that the deracemization step proceeds well with each new ligand, although the amount of time required for complete equilibration to a single diastereomer was varied.
Figure 1
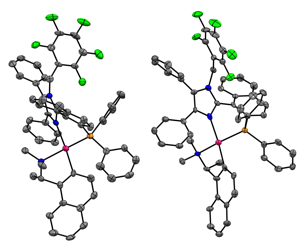
In an attempt to explain the factors responsible for the energy difference that permits equilibration to a single diastereomer, the Pd(II) complex of the rac-StackPhos¥chiral palladcycle was obtained and it was found that the diastereomers co-crystallize (Figure 2, counterions omitted for clarity). Our analysis indicates that the key difference is that, in the more stable diastereomer, the ring formed between the atoms in the ligand and the Pd (II) adopt an envelope-like conformation; whereas, in the less stable diastereomers, this ring is flattened to avoid steric interactions- likely causing it to be of higher energy and therefore less stable. A full article on this and the underlying physical organic chemistry of model systems that describes the important factors to obtain stabilization by arene arene interactions in this system has been submitted for publication and is currently under review.
Figure 2
Progress towards Aim 2:
We have shown that these ligands work well for the A3 coupling reaction, and are perhaps the most useful ligand for this reaction class. During this budget period, we attempted to merge the enantioselective alkyne addition with our Au-catalyzed heterocycle synthesis to prepare chiral 2-alkylaminoheterocycles (Scheme 2). Interestingly, when another stereocenter was present, StackPhos did not function all that well. To address with, we applied developed new ligands. PHOX and PHIM ligands have a storied history of success in catalytic enantioselective transformations and an extremely large number of congeners have been prepared in attempts to optimize selectivity for a variety of transformations. We applied our design concept to introduce axial chirality to these molecules and demonstrated that controlling this somewhat obscured element of chirality can have extremely beneficial results with respect to stereoselectivity and, in this case, outperform StackPhos with respect to catalyst control (Scheme 2). With respect to the products formed here, our goal was to use an asymmetric alkynylation/cyclization sequence to prepare 2-alkylamino heterocycles such as 5 because this important motif is found in a variety of biologically active natural products and lead compounds. The data in Table 1 indicates that the sense of chirality observed in the products is influenced by the central chirality in the backbone (entry 4), but the axial chirality can overcome this bias to provide the opposite stereoisomer (4' vs 4, entry 4 vs 2) or enhance the selectivity (entry 3 vs 4). Furthermore, these results demonstrate the potential importance of the strategy developed here, which should find widespread application in a variety of ligand types and have a far reaching impact on future applications. A manuscript describing this chemistry has been submitted for publication and is currently under review.
Scheme 2

During the last budget period, we described an efficient dearomative alkyne addition to in situ generated quinolinium salts (published in 2015), demonstrating that the catalysts can function quite well in other alkyne addition reactions. During this budget period, we have been working towards developing other reactions. Unpublished studies from our laboratory have shown that oxonium ions also undergo alkynylation reactions to provide the products in high enantiomeric excess (Scheme 3). Here the oxonium ions are formed by addition of TMSOTf to chromones. The reaction functions extremely well over a broad scope of alkynes, delivering the products nearly 90% ee or higher in most cases. We are currently working on varying the chromone moiety to study the scope here, most specifically how the electronic effects imparted by the substituents affect the ee's of the reaction. This work in nearing completion in our laboratory and will be communicated in the near future.
Scheme 3

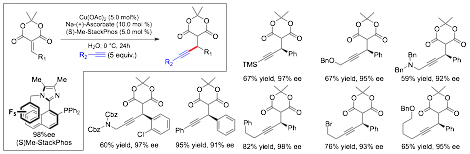
Additionally, we have been working on C-C bond forming reactions that can be used to bring together fragments in a convergent fashion, which are highly valuable transformations for organic synthesis. In this regard, we have been investigating the enantioselective addition of alkynes to Meldrum's acid derivatives. As illustrated in Scheme 4, using the methyl version of our ligand- which performs better than the parent phenyl version here- we were able to add alkynes to this class of electrophile and obtain the products in high ee's across a wide range of substrates. It should be noted that he products here are highly versatile and can be transformed into a variety of different types useful compounds. This work in nearing completion in our laboratory and will be communicated in the near future.
Scheme 4
In our laboratory, the processes of exploring additional new reactions and ligands is underway. We feel that we have only begun to scratch the surface of the new chemistry to be developed here. This ND grant has allowed us to open up a new area of research and it made a large impact on the direction of my group, likely for at least the next 5 years.










