Reports: UR353707-UR3: Synthesis and Reactivity of Molybdenum and Tungsten Carbon Dioxide Complexes
 printer friendly
printer friendlySince CO2 can be activated by coordination to a transition metal, the preparation and study of isolable CO2 complexes is of great interest. Side-on (η2) coordination is often observed for monomeric complexes of CO2 which are most frequently prepared by substitution of a labile ligand. However, η2-CO2 complexes can sometimes be prepared by the oxidation of a carbonyl ligand. Here we report that a range of η2-CO2 molybdenum complexes can be prepared from TpRMo(NO)(L)(CO) by oxidation of the carbonyl ligand.
A carbonyl ligand can be thermally liberated from TpRMo(NO)(CO)2 (TpR = Tp, tris(pyrazolyl)borate or Tp*, tris(3,5-dimethylpyrazolyl)borate) in the presence of a σ-donor ligand (L) to give TpRMo(NO)(L)(CO) (Scheme 1, step 1). Using this method, thirteen complexes featuring L = P(OMe)3, PPh3, PMe3, 3-fluoropyridine (3-F-Py), pyridine (Py), 4-dimethylaminopyridine (4-DMAP), 1-methylimidazole (1-MeIm), 1,3-dimethylimidazol-2-ylidene (NHC), have been prepared and purified by column chromatography. Several of these complexes are air sensitive. In some cases, IR spectroscopy indicates that small quantities of η2-CO2 complex are formed during heating if oxygen is not rigorously excluded. Consequently, the best results were obtained by performing thermolysis in a sealed pressure tube.
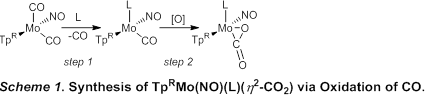
The range TpRMo(NO)(L)(CO) is constrained by the steric and electronic properties of L. For Tp, complexes featuring alkyl phosphines larger than PMe3 have not been isolated, including PEt3 and P(n-Bu)3. For Tp*, the more sterically demanding ligand platform likely prevents isolation of PPh3 and NHC complexes. On the other hand, the added electron-donating ability of Tp* allows a 3-fluoropyridine (3-F-Py) complex to be isolated for Tp*, while the analogous Tp version cannot be isolated. This result implies that π-acceptance may be more important that σ-donation for the 3-F-Py ligand; consequently, the more electron rich Tp* complex is isolable while the less electron rich Tp congener is not.
Oxidation of a carbonyl complex to form an η2-CO2 complex can be accomplished using either a hydroperoxide or oxygen gas (Scheme 1, step 2). Most commonly a hydroperoxide gives the best yields of η2-CO2 complex. Excess hydroperoxide is always required to ensure complete reaction of the carbonyl complex. Thus far, η2-CO2 complexes are isolable for a range of complexes featuring σ-donors that are phosphines, pyridines, imidazoles, and N-heterocyclic carbenes (Table 1). Carbon dioxide complexes cannot be isolated for the more electron deficient phosphines (L= PPh3, P(OMe)3). For these complexes, cumene hydroperoxide does not oxidize the requisite complex. Stronger oxidants such as PhIO do react, but CO2 complexes cannot be isolated or definitively identified in situ via spectroscopic methods. It seems likely that a CO2 complex is formed under these conditions but that the resulting complex is not stable under such harshly oxidizing conditions. This could be due to the complex becoming too electron deficient to maintain coordination of CO2. Alternately, the putative η2-CO2 ligand could be further oxidized (to a carbonate, for example), or the metal complex could be oxidized upon liberation of CO2 gas. Thus far, the least electron rich η2-CO2 complex isolated is TpMo(NO)(PMe3)(η2-CO2) based on IR spectroscopy and oxidation potential as measured by cyclic voltammetry.
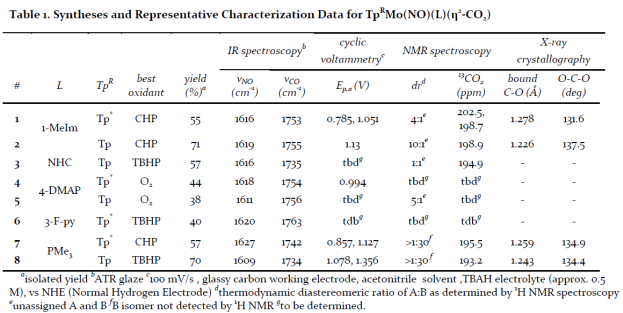
Figure 1 shows the IR stretching frequencies of all thirteen TpRMo(NO)(L)(CO) complexes displayed as a correlation of carbonyl versus nitrosyl stretching frequencies. As expected, since both stretches indicate the electron density at the metal center a linear trend is seen with a slope of one. Unsurprisingly, carbonyl and nitrosyl stretching frequencies are excellent indicators of the electron density of TpRMo(NO)(L)(CO) complexes. Complexes featuring νCO > 1893 cm-1 and νNO > 1607 cm-1 did not result in isolated η2-CO2 complexes upon oxidation. However, all isolated TpRMo(NO)(L)(CO) complexes featuring νCO and νNO lower than these have led to isolable η2-CO2 complexes upon oxidation.
The η2-CO2 ligand is often quite labile; indeed thermal instability is one reason why carbon dioxide complexes have been difficult to isolate. Furthermore, molybdenum η2-CO2 complexes have been shown to be prone to disproportionation. In contrast, 1-8 are thermally stable in solution at room temperature for days. For example, when TpMo(NO)(NHC)(η2-CO2) 3, is dissolved in DMSO-d6, heated at 50 °C and monitored by 1H NMR spectroscopy, no decomposition is observed over 4 d.
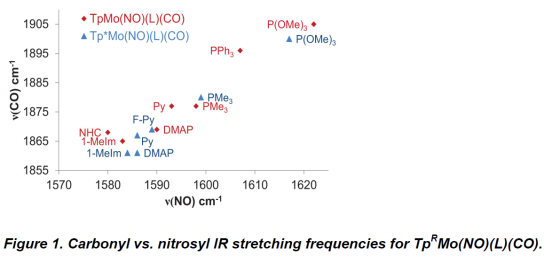
Single-crystal X-ray crystallographic analysis has been successfully performed for a number of these new η2-CO2 complexes. For previously reported η2-CO2 complexes of this type, such as TpW(NO)(PMe3)(η2-CO2), two types of positional disorder complicate the analysis of the diffraction data. First, the two η2-CO2 coordination diastereomers co-crystalize making the coordinated C=O atoms of the CO2 ligand difficult to model separately. Second, the two enantiomeric forms are also present and cause positional disorder between the uncoordinated CO of η2-CO2 ligand and the NO ligand. Fortunately the X-ray diffraction data for the analogous molybdenum complexes that feature L = PMe3 (7 and 8) are less problematic. Only one diastereomer, in which the uncoordinated oxygen of the η2-CO2 ligand is proximal the PMe3, is isolated as it is the thermodynamically-favored coordination diastereomer. Consequently, neither set of diffraction data features disorder due to coordination diastereomers. In addition, complex 7 crystalizes as a single enantiomeric form which eliminates such positional disorder from the diffraction data.
Figure 2 compares the average bond lengths and bond angle for the η2-CO2 ligand for all three TpRM(NO)(PMe3)(η2-CO2) complexes. As expected, the C-O bonds lengthen as the complex becomes more electron rich. The more donating Tp* gives rise to longer CO2 bonds, but the effect is not as dramatic as changing the metal from molybdenum to tungsten. The tungsten complex also shows a more acute O-C-O bond angle, a clear indication of stronger backbonding into the π* orbitals of the η2-CO2 ligand.