Reports: DNI453824-DNI4: Electrostatically Tethered Catalysts for the Rate Acceleration of H-bond Mediated Transformations
 printer friendly
printer friendly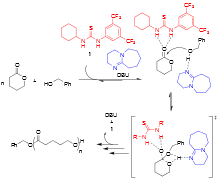
Figure 1. Accepted mechanism for the H-bond mediated ROP of lactones.
The proposed research activities were centered around the study of cocatalytic transformations and the non-covalent interactions of the cocatalysts with the broadly-defined goal of controlling these secondary interactions to alter their reactivity. H-bonding catalysts were chosen because of the diversity of chemical transformations the can be executed by their application. The chemical reaction used to study these systems was ring-opening polymerization (ROP), a field which has an established history of using H-bond mediated, organocatalytic, transformations. Through the course of studies, it was determined that in addition to moderate binding between H-bond donating thiourea (TU, 1 in Figure 1) catalysts and substrates, there exist strong binding between the cocatalysts. This latter binding energetically dominates during the course of the reaction. A base cocatalyst is required for these transformations, and it serves to activate the propagating alcohol, Figure 1.
Thiourea/amine base cocatalysts are commonly employed for well-controlled, “living” organocatalytic ROPs of cyclic esters and carbonates. In the first publication resulting from PRF support, several of the most active cocatalyst pairs were shown by 1H NMR binding studies to be highly associated in solution, dominating all other known noncovalent catalyst/reagent interactions during ROP. One strongly binding catalyst pair behaves kinetically as a unimolecular catalyst species, which accounts for the high activity of the bimolecular catalyst system. The strong binding between the two cocatalysts accounts for the selectivity exhibited by them in ROP by limiting deleterious side reactions. The predictive utility of these binding parameters was shown to be applicable to the discovery of new, highly active cocatalysts. This study was extended to thiourea/weak base (alkylamine) cocatalysts, which exhibit much weaker binding constants to each other vs TU/amidine bases. The nature of these cocatalyst interactions vary with the identity of the alkylamine, and kinetic analyses of the organocatalytic ROP reactions revealed noninhibitory behavior in [alkylamine] and a second order mode of activity for thiourea. The observation of a second order reaction directly led to the synthesis in our lab of some of the most highly active H-bond mediate catalysts known.
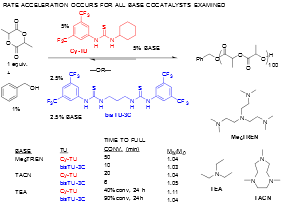
Figure 2. Rate acceleration observed for bisthioureas.
A cocatalyst system consisting of an alkylamine base and a bis(thiourea) featuring a linear alkane tether was shown to dramatically increase the rate of ROP of L-lactide (LA) versus previously disclosed monothiourea H-bond donors, Figure 2. The rate acceleration occurred regardless of the identity of the alkylamine cocatalyst, but the ROP remained controlled. This H-bond mediated ROP of LA constitutes a rare, clear example of rate acceleration with bis(thiourea) H-bond donors versus monothioureas, and the bis(thiourea) is shown to remain highly active for ROP at fractional percent catalyst loadings. These enhanced rates have since been shown to occur for other lactone monomers. A proposed mechanism for the ROP reaction with our new H-bonding catalysis is proposed in Figure 3.
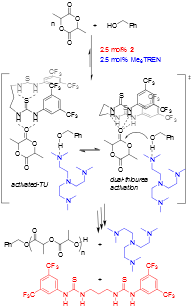
Figure 3. Proposed mechanism for bisTU cocatalyzed ROP.
Work that is still ongoing asks the fundamental question of how altering the electronic properties of the H-bond donating catalyst effects the transformation. The Lewis acidity of the H-bond donating catalyst simultaneously alters the energetics of the reactant and location of the transition state in the reaction coordinate knowledge of which we anticipate will inform future generations of catalyst. The catalyst electronics simultaneously modulate binding to substrate and to cocatalyst, and understanding the relative binding ability to reactant and cocatalyst will allow the community to devise systems with improved selectivity and activity. Collectively, these studies are elucidating a level of reaction control rendered by H-bond donating moieties that is unprecedented in organic synthesis.
This study on structurally similar catalysts is being extended to a structurally-diverse series of H-bond donating catalysts. By comparing a series of H-bond donating catalysts, most of which were first synthesized in our lab, we have discovered that the most active H-bond donating catalysts possess a certain ratio of binding constant to cocatalyst to binding constant to substrate. Maintaining this ‘ideal’ binding constant, we hypothesize, allows the catalyst to remain active despite inhibitory interactions between the cocatalysts. These studies will also allow us to explain how we are able to increase both the activity AND selectivity of our catalysts.
The work funded by the PRF has supported 6 graduate students and 3 undergraduate researchers over the course of the studies. Three of these graduate students have used their PRF-funded work as the basis for their Comprehensive Examinations, which are done as part of their graduate requirements at the University of Rhode Island (URI). Graduate students have used PRF support to travel to scientific conferences and present the results of their work. We have established scientific collaborations as a direct result of these meetings, and we are currently in the process of applying for funding based on these nascent collaborations.
The support provided by PRF has been instrumental to the PI’s career. He used initial results from the PRF-funded research as the basis for a successful NSF CAREER proposal. One of the graduate students supported by the PRF was awarded a research fellowship after a university-wide competition; his PRF-funded work was instrumental in convincing the selection committee of his ability to conduct top tier research. The PI was also recognized with a university-wide award based on research accomplishments. The publications supported by the PRF were specifically mentioned in the award citation, only one of which is given each year. Last, PRF support has been instrumental in establishing the career of a tremendously-promising undergraduate researcher. Funds provided by the PRF allowed an undergraduate researcher in the PI’s lab to quit his part-time job so that he could focus on research year-round. The results have been simply spectacular. He has two first author publications in ACS journals, and he was awarded a Goldwater Scholarship in 2016. He was the only Rhode Islander to receive a Goldwater Scholarship this year. He is currently finishing his senior year, and he will be applying to graduate schools this fall. PRF support has undoubtedly changed the life trajectory of this astonishingly-bright undergraduate student.










