Reports: ND1054075-ND10: Designing Lithium Based Hard Materials from First Principles
 printer friendly
printer friendlyLi-Mg binaries:
High strength, lightweight, low-density and good stiffness are the key properties of materials that are highly desirable in industries. Amongst all possible candidate elements, Mg stands as one of the most promising candidates. Mg is one of the lightest elements with many features that are highly desirable. However, this element also shows many drawbacks that limit its applications in the current technologies. One way to improve the mechanical properties of Mg is through alloying. We notice that the low ductility of Mg is due to its hexagonal crystal structure. However, structures with cubic symmetry are known to exhibit good mechanical properties. Therefore, we aim to search for stable cubic structures of Mg-based alloys. We start alloying Mg with Li which is the lightest solid-element present on earth. We use the minima hopping method to explore the potential energy surface (PES) of Mg1-xLix (0<x<1) and search all the chemically stable structures for different Li-concentrations. Left figure 1 shows the calculated convex-hull of Li-Mg binary system. Interestingly, we did find some low-energy structures of Li-Mg having cubic symmetry and good mechanical properties. We recently published our results on properties of Li-Mg binary systems in the J. Alloys Comp.
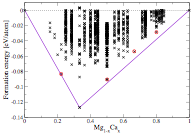
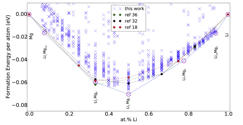
Figure 1: Left: Convex hull of the Li-Mg alloy. Right: Convex hull of the Mg-Ca binary system.
Mg-Ca binaries:
In addition to the Li-Mg binaries, we have also investigated the effect of Ca-concentrations on the physical properties of Mg-Ca binaries. The good oxidation and combustion resistance of Mg-Ca alloys makes it very attractive for technological applications. The reported theoretical and experimental results show that the most commonly known Mg-Ca alloy is Mg2Ca which has C14 leave phase in P63/mmc (194) space group. We have studied the PES of Mg-Ca compounds by using the Firefly method (FF) as implemented in our PyChemia package. Forces, energy and stresses are evaluated by using a method based on Artificial Neural Networks (NNs), as implemented in the MAISE code. Our calculations on Mg-Ca system reveal that several compositions of Mg-Ca binaries are chemically and thermodynamically stable. Our value agrees very well with previous theoretical calculations. We find three other possible stable structures that are very close to the theoretical convex-hull (right fig. 1). Presently, we are investigating the other physical properties of the obtained stable structures of Mg-Ca binaries.
Ni-Ti binaries:
The shape-memory effect of the equiatomic Ni and Ti alloy is a widely known and studied effect that is believed to rise from a martensitic transformation from a cubic to an orthorhombic structure. However, other than the equiatomic composition, little is known about other possible compositions of Ni and Ti. Therefore, the interest is not only to find the lowest energy configuration but to find those which can be chemically synthesizable. In that respect, we investigate the PES of NixTi1-x (0<x<1) and study the physically properties of the stable structures Fig-2(i).
We find all known structures and additionally also discovered a totally new structure (figure-2(ii)), Ni5Ti1. We have assessed the thermodynamic and elastic stability of these low-energy structures (figure 2(ii)). For the most widely studied equiatomic composition, our results are in good agreement with previous results.
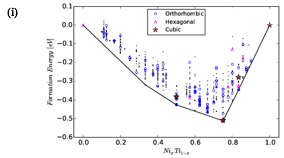
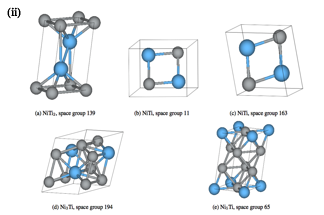
Figure 2: (i) Ni-Ti Calculated convex-hull. (ii) Selected low-energy structures.
Bi-Sb binaries:
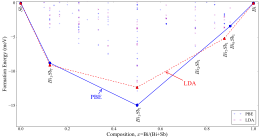
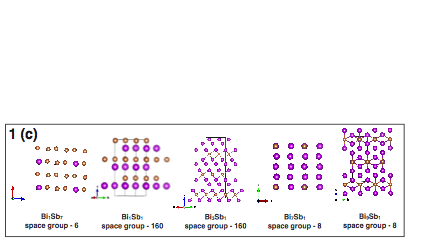
These alloys recently attracted lot of attention due to their numerous applications in the spintronics and thermoelectric industry. Even though, these alloys were known since last few decades, the complete phase-diagram of Bi-Sb binary system has been lacking in literature. We have employed the minima hopping structural search method to investigate the full phase-diagram of Bi-Sb alloys at zero temperature and pressure. We found several new chemically stable structures together with many metastable structures, which could be experimentally synthesized (figure-3).
We have obtained that Bi1Sb1 (space group 160) has the lowest formation energy. Interestingly, we notice that by applying pressure on Bi1Sb1 compound, one can realize a Weyl semi-metallic phase in this system. Results that have been accepted for publication in Physical Review B (Rapid Communications). Additionally, we have systematically studied the properties of all possible-stable and few-metastable structures. We found the presence of novel electronic properties such as 3D topological insulator, Weyl semimetal, giant Rashba effect and good thermoelectric properties in some of the studied Bi-Sb systems.
![]()
![]()
Figure 3: (i) Calculated Bi-Sb convex-hull. (ii) Some predicted low-energy crystal structures,.
Li-Au binaries:
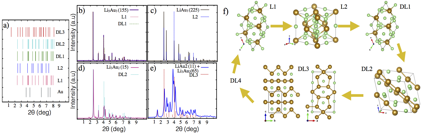
Recently, Li-Au alloys have been attracted great deal of attention from its potential application on anode in Li-ion batteries. In order to understand the process of lithiation in Li-Au alloys, we have with an experimental group headed by prof. Frank Renner at Hasselt University in Belgium. In such, we have performed an experimental and theoretical structural characterization during electrochemical process of Li-Au. A correspondence of the experimental structures obtained during the electrochemical experiments has been done using the Minima Hopping method to determinate the observed structures. A comparison of simulated X-ray diffraction patterns with experimental measurements is detailed described. A summary of our findings is shown in Figure 4. For phases L1 and DL1 correspond to Li5Au3 crystal structure with space group R32 (155). For phase L2 we found that Li3Au with space group Fm-3m (225) matches the predicted experimental XRD. For phase DL2 a Li3Au2 phase with space group C12/c1 (15) is proposed. In phase DL3, we found a combination of two phases, which consist on 90% of Li3Au5 with space group Cmmm (65) and 10 % of LiAu2 with space group P121/m1 (11). The simulated XRD is in reasonable agreement with experimental X-ray diffraction pattern. Figure 4f shows the atomic arrangement for the structures proposed. It is important to mention that this work have been published in Chemistry of Materials.