Reports: ND351566-ND3: Electrochemically-Controlled Alkene Binding Affinity of Surface-Immobilized Metal-Thiolate Complexes
 printer friendly
printer friendlyIn collaboration with Professor Hao Chen of Ohio University and Dr. Larry Campbell of AB Sciex, we reported the gas phase reactivity of our metal-stabilized thiyl complexes. This work supports our hypothesis that the alkene addition reaction involves metal-stabilized thiyl radical intermediates, a new type of distonic ion. The addition reactions of [Ru-1]+ with alkenes or methyl ketones in the gas phase were observed in agreement with the proposed mechanism. Such reactivity is also maintained by several fragment ions of [Ru-1]+, indicating that the preserved thiyl diradical core structure is responsible for the addition reaction. The thiyl radical nature of [Ru-1]+ was further verified by the ion/molecule reaction of [Ru-1]+ with dimethyl disulfide, in which the characteristic CH3S• transfer occurs, both at atmospheric pressure and also at low pressure (~mTorr). These results provide, for the first time, clear mass spectrometric evidence of the radical nature of [Ru-1]+ arising from the oxidation of non-innocent thiolate ligands of the complex [PPN][Ru-1]. The results provide support for our reaction method and an alternate method to selectively detect and identify alkenes at low concentrations by mass-spectrometry.
The addition of polysubstituted alkenes, alkynes, and dienes to the metal-stabilized thiyl radical complex [Ru-1]+ was evaluated by electrochemical, chemical, and computational methods. Rate constants for polysubstituted alkenes range from 1.1 × 101 to 8.8 × 102 M-1 s-1, which are at 3–5 orders of magnitude slower than their monosubstituted counterparts. Rate constants are slower for substituted alkynes varying from 2.3 × 102 to 1.2 × 104 M-1 s-1, which are ∼100 times slower than the corresponding alkenes. These results help to establish the kinetic selectivity of our system for ethylene over other alkenes.
Studies of the oxidative degradation of our metal-thiolate complexes revealed new crystal morphology of the sulfur-oxidized product. The x-ray crystal structure allows the unambiguous structural confirmation of a protonated sulfenic acid donor to a metal, which represents the first M-S(OH)R structure with refined H-atom coordinates/thermal parameters. Density functional theory computations confirm the ligand protonation state and metal oxidation state.
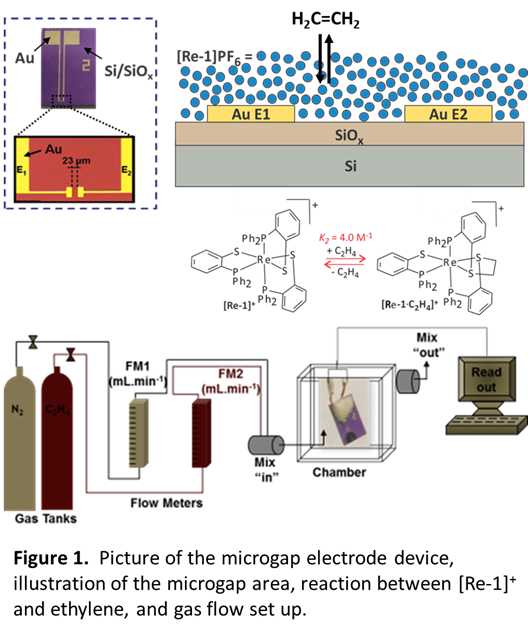
The electronic conductivity of solid-state films of the [Re-1]+ complex changes in the absence and presence of ethylene. A film of the metal complex was deposited by dropcast across a microgap electrode of two Au electrodes separated by 23 μm that were fabricated in a clean room facility by photolithography on a Si/SiOx substrate (Figure 1). Wire leads were attached to the Au contact pads and the electrodes were thoroughly cleaned. A 3 mM [Re-1]+ complex solution was prepared by oxidation of [Re-1] with ferrocenium hexafluorophosphate in dichloromethane inside a nitrogen filled glovebox. The deep blue solution of [Re-1]+ was transferred dropwise to the electrode surface and allowed to evaporate forming a thin film.
The radical complex [Re-1]+ bridges the gap between the Au electrodes (Figure 1). The modified electrode was transferred to a gas mixing chamber with ethylene and nitrogen supplies (Figure 1). A bias of 1 V was applied and the current measured in the presence of variable ethylene: nitrogen ratios under ambient conditions. Exposure of the [Re-1]+ modified electrode to 100% ethylene with an applied bias of 1 V results in a significant decrease in current (Figure 2). Bond formation between [Re-1]+ and ethylene yields the metal-centered radical product [Re-1·C2H4]+, which quenches the sulfur-centered radical character, resulting in increased resistivity. The current is restored when nitrogen is reintroduced. Repeated cycling of 100% ethylene and 100% nitrogen confirms the reversibility of ethylene binding and reproducibility of the current response (Figure 2).
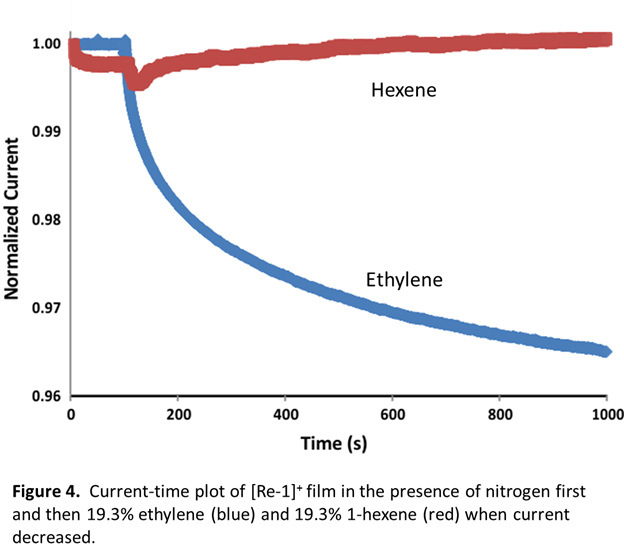
Figure 3 shows the response of the film to ethylene concentrations between 0.9% and 31.3%. The average percent response is 0.41-3.52 with standard deviation from 0.06- 0.39. The response increases with increasing ethylene concentration until the binding sites within the film become saturated and the response begins to plateau near 30% ethylene (Figure 3). The response is highly stable and selective towards ethylene. Electrodes stored for up to 6 months under ambient conditions showed no significant change in response to 100% ethylene despite the air-sensitivity of [Re-1]n in solution. The selectivity of the sensor derives from the steric constraint of the ethylene binding site as confirmed by prior solution studies. To demonstrate the selectivity, the film was exposed to a gaseous mixture of 19.3% 1-hexene and nitrogen with no detectable change in resistivity compared to the response to the same concentration of ethylene (Figure 4).
Our results show that the [Re-1]+ complex reacts selectively with ethylene similarly in solution and in the gas phase. Using simple dropcast methods, we monitored the reversible binding of gaseous ethylene to the solid [Re-1]+ complex. The conductivity change in the film is attributed to hindered electron hopping in the presence of ethylene due to localization of the electron spin on the metal-center as opposed to the electrons being delocalized in the ethylene-free complex. The selectivity is similar in the solution and gas phase, showing similar reactivity in both media. The selective and reversible nature of this system is unique. These results were recently submitted for publication to RSC Adv. Future studies will be aimed at studying the solid-state reactivity of ethylene with the stronger binding [Re-1]2+ complex, which has a binding constant of 2.5 x 109 M-1.