Reports: DNI651680-DNI6: Accurate Ab Initio Studies of Hydrocarbon Physisorption on Carbon Nanotubes
 printer friendly
printer friendlyDuring the initial twenty months of the PRF project, the research was carried out by Daniel Smith, a graduate student currently in his third year, under the guidance of the PI. At this stage we have focused on three goals:
1. to obtain high-level benchmark interaction energies for model systems representing methane adsorption on graphene sheets and carbon nanotubes of different curvature,
2. to assess the accuracy of various novel density functional theory (DFT) variants relative to benchmark interaction energies,
3. to utilize the top-performing DFT variants to find out minimum-sized fragments that lead to converged binding energies and to study the dependence of adsorption energy on the nanotube curvature.
The above goals have been fulfilled and the pertinent details are contained in two papers: one published and one submitted. The research relevant to this project was also presented at six conferences.
1. Model systems and accurate benchmark interaction energies
At the first stage, we selected a set of weakly interacting dimers of methane with polycyclic aromatic hydrocarbons (PAHs) benzene through coronene as model systems. For each dimer, we considered all three coordinations of the methane molecule, that is, geometries in which one (1C), two (2C), or three (3C) methane hydrogens are closer to the PAH plane than the methane carbon (Fig. 1). Subsequently, we extended the coronene and larger models to include curvature by cutting out fragments from several armchair (n,n) and zigzag (n,0) nanotubes. Benchmark interaction energies were computed for one-dimensional radial cuts passing through the lowest-energy structures at each coordination.
Following the standard practice, benchmark interaction energies were calculated as sums of the MP2 contribution extrapolated to the complete basis set (CBS) limit and the ΔCCSD(T) correction for effects beyond MP2 computed in moderate basis sets. The MP2/CBS values obtained in this work mostly utilize the (aug-cc-pVTZ,aug-cc-pVQZ) extrapolations and are converged to within a few thousandths of a kcal/mol. Similar precision is not attainable for ΔCCSD(T). However, our extensive studies for benzene-methane and naphthalene-methane, involving basis sets up to aug-cc-pVQZ and the explicitly correlated CCSD(T)-F12 approach, indicate that the benchmark interaction energies are accurate to within 0.1 kcal/mol or better even for larger dimers where the ΔCCSD(T) term was computed in the aug-cc-pVDZ basis set (somewhat reduced for coronene-sized models to avoid CCSD convergence problems).
While the benzene-methane dimer, with its 1C minimum structure, has been extensively studied before, the interaction energies for larger complexes have not been previously obtained with any comparable accuracy. Our results in Fig. 2 indicate that, for all larger dimers, the global minima are triply coordinated. This remains true for curved PAHs, however, for the inside adsorption on nanotubes about 7-8 in diameter, the differences between coordinations nearly vanish as the adsorbed molecule is virtually on the nanotube symmetry axis. This inside configuration is actually the most favorable one with binding energies approaching 10 kcal/mol.
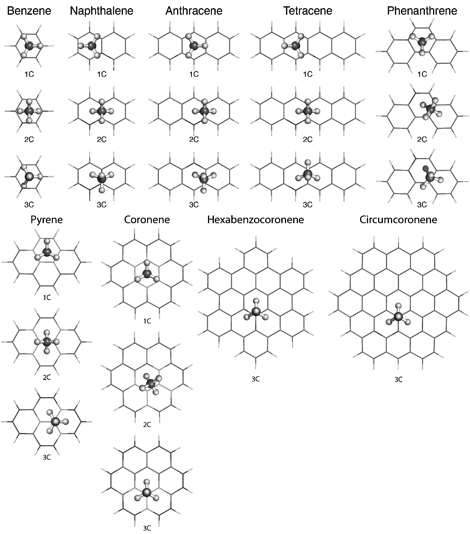
Figure 1. Lowest-energy structures for the singly- (1C), doubly- (2C), and triply-coordinated (3C) methane-PAH complexes considered here.
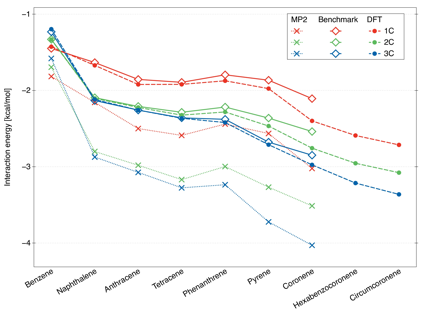
Figure 2. Interaction energies for the lowest-energy 1C, 2C, and 3C structures of different methane-PAH dimers. The benchmark values are obtained as CBS-extrapolated MP2 energies augmented by the ΔCCSD(T) correction. The values marked as DFT denote the B3LYP-D3/aug-cc-pVDZ functional.
2. Selection of the optimal DFT variant
It is well documented that standard density functionals do not reliably treat the dispersion interaction, which is the main binding force between methane and finite or infinite carbon nanostructures. Therefore, one needs to resort to novel DFT approaches that have been formulated with weak interactions in mind. In this work we tested two classes of such approaches:
1. the DFT+D method using standard B3LYP, B97, and PBE functionals augmented by the –D2 and –D3 empirical dispersion expressions,
2. functionals that have been explicitly optimized for weak interaction energies: M05-2X, M06-2X, and ωB97X-D.
The above DFT approaches in several basis sets were employed to compute methane-PAH and methane-curved coronene interaction energies along the selected one-dimensional potential cuts. Compared to the CCSD(T)-level benchmark, the interaction-optimized functionals perform quite poorly – none of them gets below 10% in median relative accuracy. On the other hand, several counterpoise-corrected DFT+D approaches are remarkably accurate. The top performers for the exterior and interior adsoprtion sites, identified using the methane-curved coronene results, are B3LYP-D3(BJ)/def2-SVP and B97-D3-E(3)/aug-cc-pVTZ, respectively, leading to a mean relative accuracy of 3.4% and 4.3%, respectively. Thus, these functionals are appropriate for studying methane adsorption on larger nanotube fragments.
3. Extended nanotube models
Equipped with an accurate and computationally efficient DFT approach, we set out to compute interaction energies between methane and larger nanotube fragments. We have investigated both circumcoronene-like models and complete nanotube fragments of finite length. We observed (Fig. 3) that for the exterior adsorption sites a circumcoronene model is virtually converged, however, for interior sites significantly larger models are needed to accurately recover the adsorption energy. Moreover, for selected nanotubes we have performed periodic plane-wave PBE-D3(BJ) calculations (using the VASP code purchased from this grant) for the largest feasible finite models (where the results were in good agreement with conventional Gaussian-basis calculations) and for infinite nanotubes. The results show that, at least at this level of theory, the finite models employed by us are fully adequate.
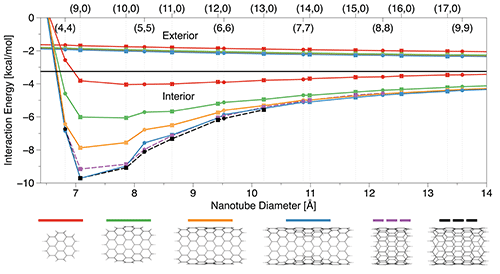
Figure 3. DFT interaction energies for the methane-nanotube fragment complexes in the 3C configuration. The black line represents the value for flat graphene.
4. Future work
Having selected the theory level and model size appropriate for studying methane-nanotube interactions, we are working on obtaining interaction energies for a large number of intermolecular geometries so that we can construct an analytical adsorption potential and employ it in simulations of adsorption isotherms and of the vibrational dynamics of methane on the surface. In addition, we are carryning out benchmark calculations for analogous models of the CO2 adsorption (which is important for the sequestration of CO2 from flue and exhaust gases). A new graduate student in my group is starting interaction energy calculations involving models of nitrogen-doped carbon nanotubes.









