Reports: ND351077-ND3: Non-Innocent Ligands as a Redox Switch for Copolymerization Reactions
 printer friendly
printer friendlyThe synthesis of new polymers is motivated by limitations of current materials. Recently, interest in polyolefin copolymers containing both semicrystalline and amorphous blocks has been increasing since it is believed that these materials could be more cost effective and have potential for further performance enhancements, allowing competition against styrene block copolymers. Living Ziegler-Natta polymerization has been used for the formation of block copolymers by sequential monomer addition, but traditional approaches are not cost effective since only one chain per catalyst molecule is generated. Several alternatives have emerged to circumvent this problem. One approach, known as chain shuttling, involves two catalysts and a transfer agent. Each catalyst polymerizes specifically one type of olefin, generating blocks that alternate automatically because the shuttling agent transfers the polymer chain to the other catalyst. However, control of both the structure and molecular weight of the resulting polymers is difficult.
In the quest of developing alternatives to metallocene a-olefin polymerization catalysts, chelating diamides have emerged as great supporting ligands for early transition metals. Such endeavors have provided an important stimulus for the synthesis of 1,1'-di(heteroatom)-functionalized ferrocenes, where the heteroatom is nitrogen, oxygen, and sulfur. The existing examples of ferrocene-based group 4 metal catalysts show poor activities towards the olefin polymerization. Based on current theories of olefin polymerization, there might be two possible problems, electronic and steric factors. For example, 1,1'-ferrocenyl diamide ligands are always bidentate, provide formally 10 electrons to the metal center, and lack protection from bulky groups. All these factors lead to highly unsaturated and active species that are unstable during the polymerization process. Another existing example, ferrocene-tethered salen ligands (salfen, Figure 1), because of their rigid coordination configuration, always position the auxiliary ligands, benzyl or amido, trans to each other. It is known that polymer chain propagation is prohibited for the group 4 metal catalysts when the vacant coordinate site is trans to the alkyl group. The phosfen ligand (Figure 1) developed by our group suffers from the same problems as the ferrocene-derivatized salen ligands.
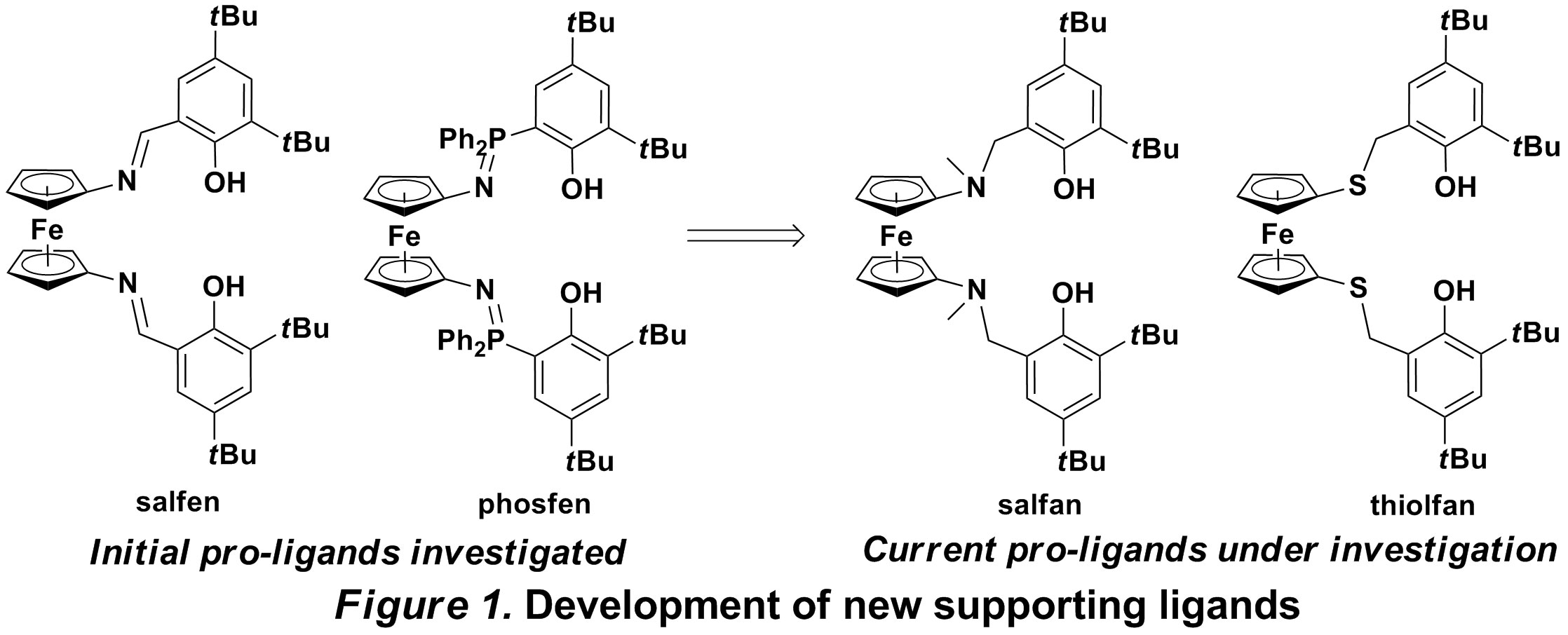
Hence, we decided to develop another system for olefin polymerization, in which the core metal is well wrapped and the auxiliary ligands are cis. At the same time, the oxidation of iron in the ferrocene-based ligand is also crucial; in the ideal situation, only one of the oxidized and reduced states would perform well toward a monomer while the other state would prove inert. Based on this assumption, such a catalytic system could be employed also as a chain shuttle agent for olefin polymerization without any additional reagents required.
Our initial efforts focused on derivatives of salan ligands (Figure 1). We believe the electronic and steric effect could be easily tuned for such ligands because different groups can be easily introduced on the phenolate part and the amine part. The target pro-ligands are prepared in high yield, and referred to as salfan (nitrogen substituent) and thiolfan (sulfur substituent). By attaching a benzyl group on nitrogen or sulfur, these pro-ligands exhibit higher air- and moisture- stability and provide a more flexible coordination environment than other 1,1'-ferrocenylene ligands. The corresponding zirconium dibenzyl complexes, (salfan)ZrBn2 and (thiolfan)ZrBn2 (Figure 2), were prepared in high yield by alkane elimination from zirconium tetrabenzyl in toluene at -78 °C. The characterization of the complexes by 1H NMR spectra indicates, as expected, C2 symmetry in solution. The oxidation of those complexes (Figure 2) was investigated by both electrochemical and chemical means. Cyclic voltammetry experiments ([nBu4N][BArF], THF, -40 °C) indicate that (salfan)ZrBn2 shows an irreversible redox event associated with the ferrocene group. Consequently, treatment of (salfan)ZrBn2 with FcBArF in toluene at -100 °C led to the oxidation of the ferrocene group, as identified by the release of ferrocene from FcBArF. Unfortunately, further studies indicated that, with time and increase in temperature, the benzyl group on zirconium was eliminated as dibenzyl, which could be attributed to the reductive nature of group 4 metal carbon bonds. Meanwhile, (salfan)ZrBn2 was also activated with B(C6F5)3 at room temperature, to give the cationic monobenzyl complex exclusively. However, polymerization of 1-hexene was not successful and only low activity was observed at room temperature.
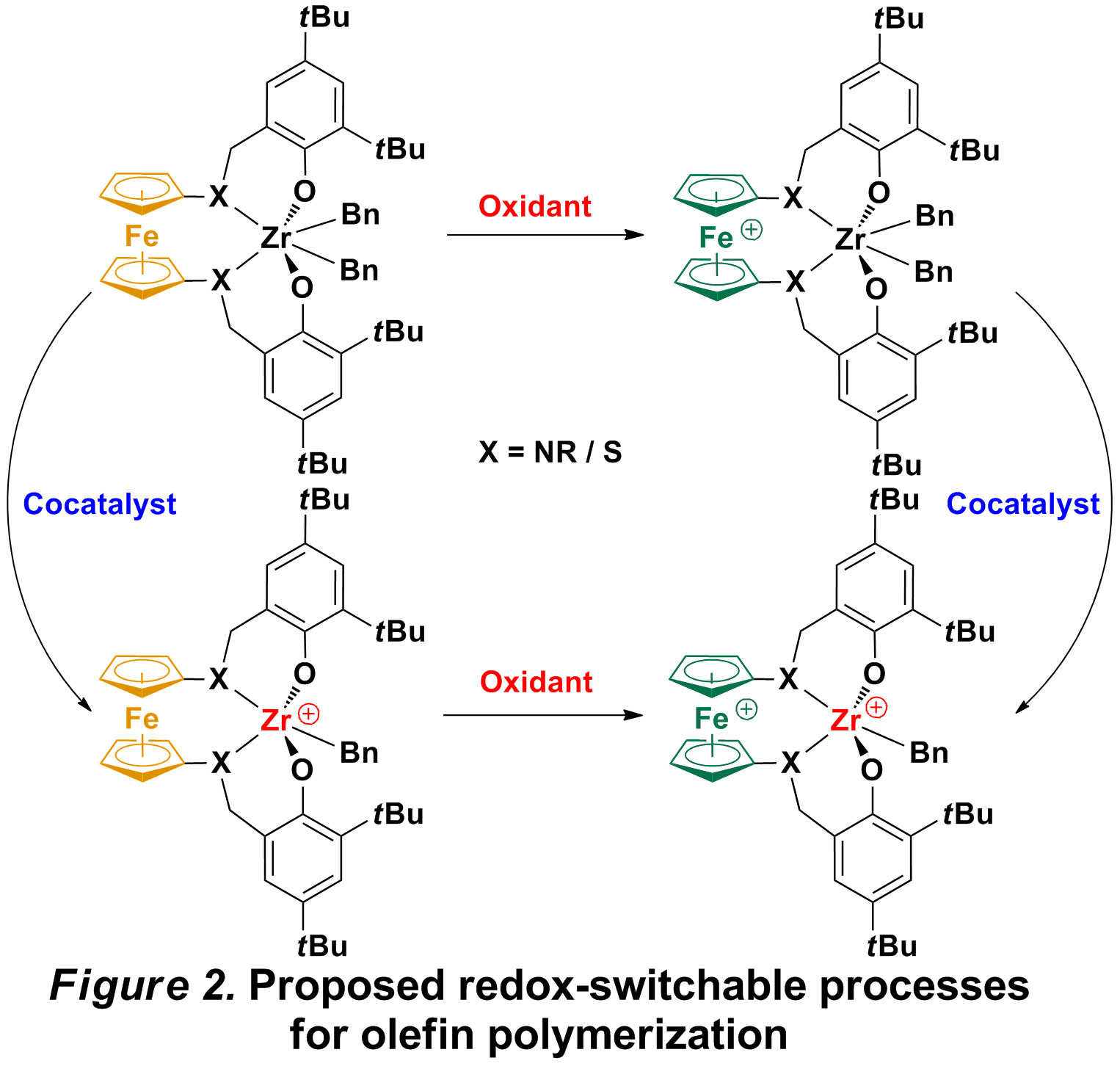
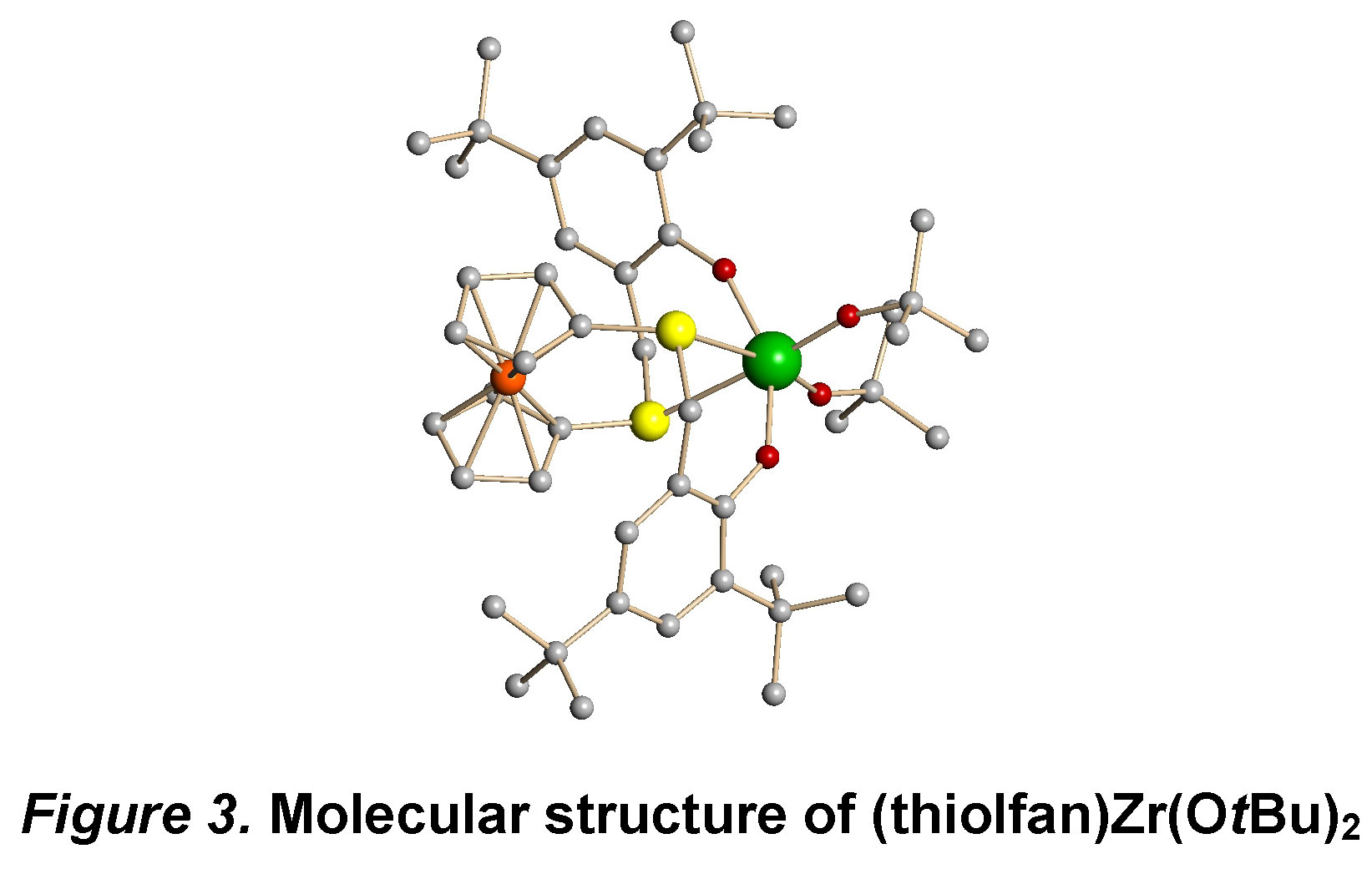
While still investigating possible variations for olefin polymerization, we decided to study other polymerization reactions with the systems described above. Since the presence of zirconium-carbon bonds could be challenging, we pursued other metal-heteroatom bonds that would be more robust under redox conditions. Consequently, complexes with Zr-O bonds were synthesized, such as (salfan)Zr(OtBu)2 and (thiolfan)Zr(OtBu)2. The C2 symmetric structures of these complexes were confirmed by both by 1H NMR spectra in solution and X-ray spectroscopy in the solid state (Figure 3). Redox switching experiments were carried out, and all the complexes could be switched fast and completely by using certain oxidants and reductants.
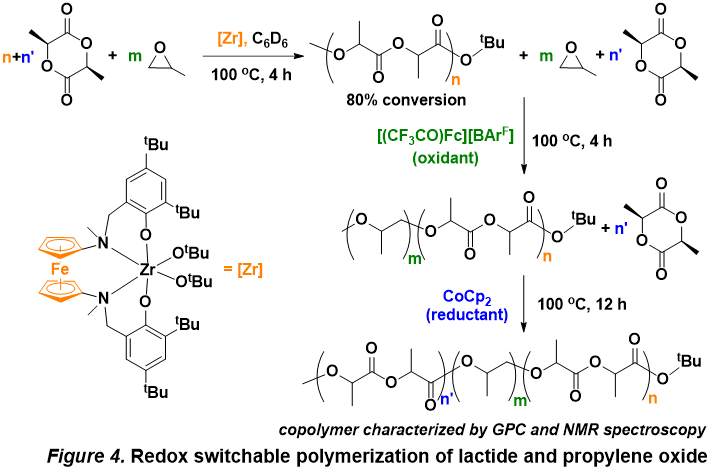
Initial studies of polymerization reactions are promising and indicate that the reduced complex initiates the polymerization of lactide, while the oxidized counterpart initiates the polymerization of epoxide (propylene oxide, cyclohexene oxide). We also observed, by 1H and 13C NMR spectroscopy, redox switchable polymerization by the controlled addition of redox reagents when the two monomers were added with the catalyst in the same flask (Figure 4). The resulting copolymers were characterized by GPC and NMR spectroscopy. It is possible that the mechanism of polymerization switches from coordination (lactide) to anionic (propylene oxide) ring opening since NMR spectroscopy indicated no stereoselectivity for the propylene oxide polymerization step.
In addition to understanding the results discussed above, other efforts focus on developing other metal catalysts and on characterizing the obtained polymers (MALDI-TOF, GPC, DSC). Importantly, these results indicate that the molecular weight, the composition, and the distribution of different monomers in the resulting block copolymer may be tuned manually, which is difficult to achieve with chain shuttling reagents. This is the first example of orthogonal reactivity discovered in the field of ring-opening polymerization of cyclic esters and ethers.
Overall, series of relatively stable pro-ligands and the corresponding zirconium dibenzyl and di-t-butoxide complexes have been synthesized and characterized, and redox-switching polymerization studies indicate promising results that are currently being explored in our lab.









