Reports: ND1051694-ND10: Temperature-Tunable and Switchable Colloidal Interactions in Multi-Component Fluids
 printer friendly
printer friendlyWe performed experiments on using the critical Casimir force (CCF) to control the nanoparticle (NP) assembly in binary liquid mixture. One graduate student, Ms. Sharmine Alam defended her Ph.D prospectus on this project in summer 2013 and was able to obtain useful data. She measured the critical temperature (Tc) of a binary liquid mixture as a function of NP volume fraction. NP chosen was Ludox particles (Sigma Aldrich, Inc.) of radius 10 nm and was used at varying volume fraction. The critical mixture was a 2, 6 lutidine + water (LW) near its critical volume fraction ratio 22:78. The LW system was a convenient choice due to its inverted coexistence curve and Tc being conveniently few degrees above the room temperature. The decrease of Tc with increasing volume fraction is clear as was shown in Fig. 1. The Tc was measured by observing the maximum of the scattering signal due to critical opalescence, which is a hallmark of systems near a critical point.
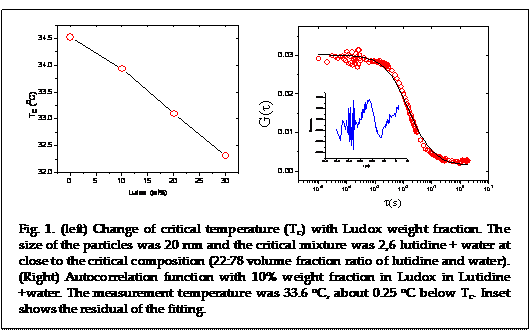
We performed fluorescence
correlation spectroscopy (FCS) experiments to measure the diffusion coefficient
(D) as a function of temperature near Tc. For these experiments a
small amount (few nM) of fluorescently labeled silica sphere is inserted into
the ternary mixture. The detailed measurements of diffusion coefficient vs.
temperature (T) are in progress. An interesting behavior was observed in the autocorrelation
function (ACF) near the critical point. Although far away from Tc,
the ACF can be fitted very well with the model of Brownian diffusion with single
diffusion coefficient close to Tc the curve deviates significantly
from the data points. The graph shown is from 30% volume fraction of Ludox
(Fig. 1 right). This volume fraction is still far away from the glass-forming
volume fraction of hard spheres (~0.58) so the data cannot be explained with
impending glass transition or caging effect. We hypothesize that this deviation
is due to attractive interaction among the particles due to the critical
Casimir force. We have estimated the center-center separation between particles
~25 nm at the Ludox concentration we used for these experiments. As the range
of CCF scales with the correlation length (ξ) which can be tens to
hundreds of nanometer close to Tc, the attractive force among the
particles will be operative. We are currently performing more systematic
measurements with different NP volume fraction to test this hypothesis.
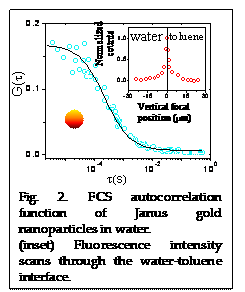
We have also successfully prepared Janus nanoparticles (JNP) and measured the autocorrelaton function. Fig. 2 shows the FCS autocorrelation function of Janus gold nanoparticles (R0~5 nm) in water prepared by single-crystal templating method (D»45 mm2/s). The sample was obtained from Prof. Chris Li, Drexel University, through research collaboration. Fluorescence intensity scans through the water-toluene interface showed much higher counts at the interface, confirming the particles' Janus character.
In parallel, the support from the PRF grant had helped us to pursue other experiments and continue students training and research. It has resulted in two publications in 2013 (Applied Physics Letters & Soft Matter) with the graduate student Ms. Indermeet Kohli as the first author, which has helped her to graduate in the summer, 2013. In the Appl. Phys. Lett. article we have used the method of FCS to study the interaction and diffusion of gold nanoparticles in a bovine serum albumin (BSA) solution. The Brownian diffusion of the NPs was altered by the adsorption of BSA. This adsorption was studied as a function of NP size and protein concentration. Our results indicated a BSA monolayer formation at the NP surface with a thickness of ≈3.8 nm. No multi-layer formation was observed even at significantly higher protein concentration. The NP diffusion was observed to follow the prediction from Stokes-Einstein relation using the bulk viscosity provided the monolayer thickness was taken into account. Figure 3a represents the measured hydrodynamic radii of 2.5 nm AuNPs plotted as a function of BSA concentration. The data is fitted using modified Langmuir equation. The best fit yields a dissociation coefficient of KD = 78.6 ± 9.5 µM and a Hill coefficient of n = 0.63 ± 0.03, which being below 1 indicates anticooperative binding. Comparison to Langmuir binding isotherm (n=1) is also shown in Fig. 3a. Dissociation constant (KD) increases with particle size indicating stronger interaction of BSA with smaller sized NPs. This would indicate the adsorption to be caused by ligand exchange reaction rather than electrostatic attraction mechanism. The maximum number of protein molecules adsorbed per 2.5 nm radius AuNP as obtained from the fit is Nmax = 8.4.
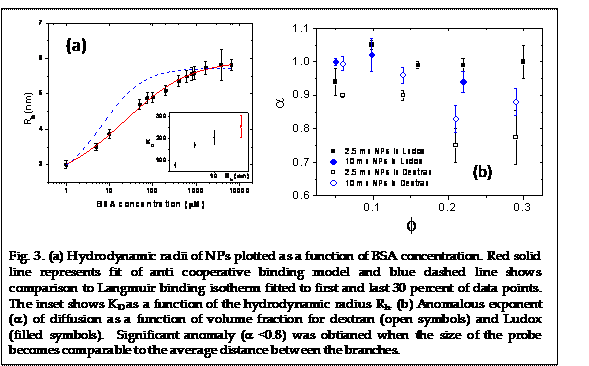
In the
other paper, Soft Matter we contrasted the diffusion of gold nanoparticles
(AuNPs) in crowded solutions of randomly branched polymer (dextran) and rigid,
spherical particles (silica). The goal was to understand the roles played by
the probe size and structure of the crowding agent in determining the probe
diffusion. Generally as the size of the diffusing species decreases their
mobility increases and as the volume fraction of the crowding agents increases
their mobility decreases. The crowding agents affect the collision frequency of
the probe particle as well as the hydrodynamic interaction and together they
influence the dynamics of the probe. We compare the probe particle diffusion in
dextran with another crowded system composed of unlabelled Ludox particles of
radius, Rp ≈10 nm. It is comparable to the radius of gyration,
Rg≈8 nm of the dextran molecules used. But in contrast to
Ludox particles, which are rigid and impenetrable spheres, dextran molecules
are soft and structured. This is reflected in differences in the probe
diffusive behavior and the rheological properties of the solution.Our results
indicated that the AuNP diffusion can be described using the bulk viscosity of
the matrix and hydrodynamically dextran behaved similar to soft colloid. In all
situations, we observed normal diffusion except for 2.5 nm sized AuNP particles
in dextran solution at higher volume fraction (Fig. 3b). This is caused by
transient trapping of particles within the random branches. The results showed
the importance of macromolecular architecture in determining the transport
properties in complex fluids.









