Reports: DNI1050934-DNI10: Effect of Surface-to-Bulk Atomic Structure on Surface Electronic Structure in Multicomponent Catalysts
 printer friendly
printer friendlyThe near-surface compositional architecture in photo, electro, and traditional heterogeneous catalyst systems fundamentally shapes their properties. Design of near surface composition often employs synthesis through solution chemistry where metal ions are reduced onto a support surface. Nominally, it is understood that as metal layers build, the reduction reaction is complete and the adlayer metal may be described by neutrally charged atoms. There is also an expected threshold lengthscale over which the support material affects the surface chemistry through electron transfers. However, for very low adlayer thicknesses of deposited species, it is not clear whether the deposit prefers to remain partially charged through effects of either low dimensionality or charge transfer with the substrate. At the theoretical
lowest-loading limit, where single atoms are deposited in monodispersed geometries and adatom–adatom distances are large enough that lateral interactions can be neglected, the tendency of an adatom to oxidize or reduce may be quite different than that of the bulk material.
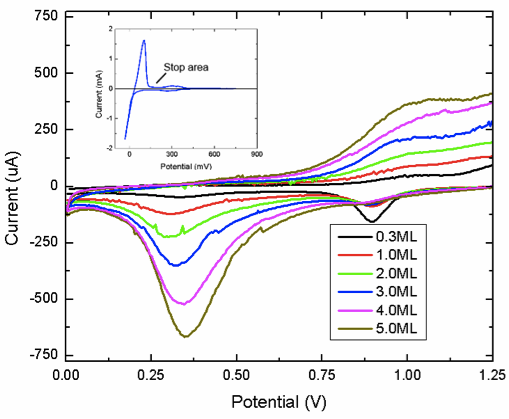
We have conducted a fundamental X-ray photoelectron spectroscopy (XPS) study of platinum-gold core-shell samples. A series of Pt-Au catalysts was prepared by Cu underpotential deposition and subsequent redox replacement of Pt4+ ions (Fig 1).
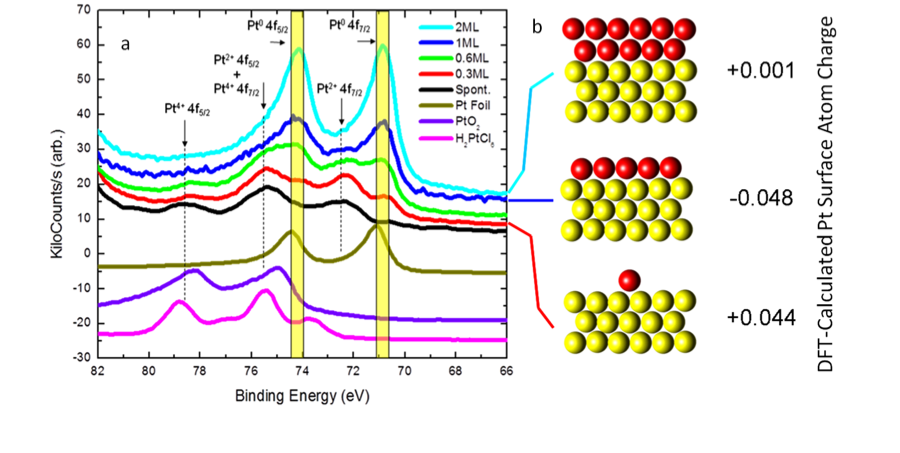
In addition to metallic Pt,
Pt2+ and Pt4+ cationic species were detected in XPS
spectra (Fig 2). DFT
analysis of the 1 ML case indicates electron transfer from gold to the first
fully formed Pt layer, followed by neutral charge for
the second layer.
 We have identified a low-loading
thickness limits of 2 monolayers below which the Pt
film exhibits decreased activity, and a limit around 3 monolayers where a
significant increase in durability can be seen. Increasing overlayer
thicknesses of Pt monolayer catalysts lead to a more
metallic surface chemistry and atomic structure in the Pt
catalyst surfaces which account for improvements in
activity and durability. Samples that were annealed showed a considerably more
metallic XPS photoemission were higher temperature annealing lead to an
alloying of the Pt overlayer
and Au substrate. Higher temperatures and annealing times lead to alloying and
to Au segregation to the surface. The electrochemical activity for oxygen
reduction reaction (ORR) showed a strong dimensional dependence. The ORR
activity showed maximum activity at 2 monolayers of Pt/Au,
with diminishing activity
(normalize to surface Pt) beyond the 2 monolayer
threshold.
We have identified a low-loading
thickness limits of 2 monolayers below which the Pt
film exhibits decreased activity, and a limit around 3 monolayers where a
significant increase in durability can be seen. Increasing overlayer
thicknesses of Pt monolayer catalysts lead to a more
metallic surface chemistry and atomic structure in the Pt
catalyst surfaces which account for improvements in
activity and durability. Samples that were annealed showed a considerably more
metallic XPS photoemission were higher temperature annealing lead to an
alloying of the Pt overlayer
and Au substrate. Higher temperatures and annealing times lead to alloying and
to Au segregation to the surface. The electrochemical activity for oxygen
reduction reaction (ORR) showed a strong dimensional dependence. The ORR
activity showed maximum activity at 2 monolayers of Pt/Au,
with diminishing activity
(normalize to surface Pt) beyond the 2 monolayer
threshold.
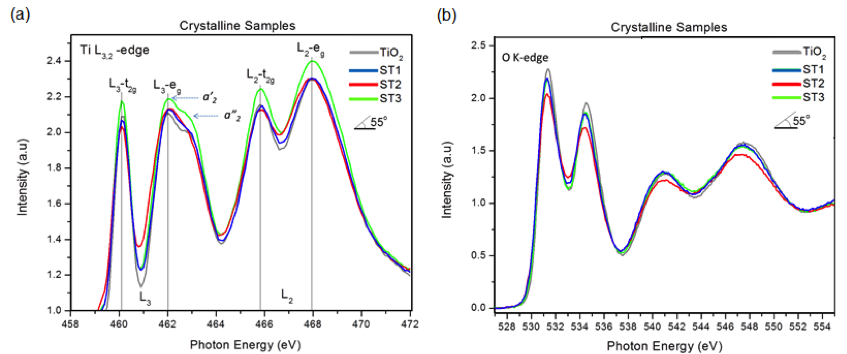
In a related study, we demonstrate the possibility to in-situ dope titania (TiO2) nanotube arrays with strontium during their synthesis. At low doping content, distinct changes in the bulk and surface structures as well as the electronic characteristics of the nanotubes were observed. Bulk analysis shows Sr to be mainly incorporated into the lattice of TiO2. Surface analysis, however, reveals phase segregation of SrO in the TiO2 matrix at high Sr doping levels. The near edge X-ray absorption fine structure (NEXAFS) spectroscopy analysis revealed that Sr2+ doping only alters the Ti and O ions interaction in the TiO2 lattice on the surface with no effect on their individual charge states. The XPS depth profile analysis reveals information about the lateral elemental distribution along the length of the nanotubes. The TEM, high resolution TEM (HRTEM) and the high angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) coupled with the electron energy loss spectroscopy (EELS) and EELS spectrum imaging (EELS-SI) measurements gives clear insights about the doping mechanism.
We found, for example, that the Ti L3,2-edge NEXAFS of the Sr-doped TiO2 and undoped TiO2 nanotube samples exhibits an intensity pattern (a2' > a2''), which is similar to that of anatase, Figure 3a. In agreement with the XRD data, this result confirms that no phase change has been occurred on the surface of Sr-doped TiO2 nanotubes after annealing. Also, the Ti L-edge with two doublet peaks is observed between 455 and 470 eV. In the TiO2 anatase structure, the Ti 2p spin-orbit interaction splits into the L3 (2p3/2) and L2 (2p1/2) levels with a separation of 5.5 eV. The L2 and L3 adsorption edges are further split into two doublet peaks of t2g and eg as sub-bands, which is due to the crystal field effect. The slight variation in the intensity and splitting in the spectra of the doped samples compared to undoped TiO2 nanotube can be related to the presence of strontium. In fact, along with the insertion of Sr2+ ion into the interstitial sites in anatase TiO2 nanotubes, electrons are doped into the lattice by filling the empty conduction bands for the charge compensation. As expected, such modification has an effect on the electronic structure of the anatase TiO2 as can be seen in the corresponding Ti L3,2-edge and O K-edge spectra. The relative intensity of the main peaks and the crystal field splitting to the t2g and eg orbitals of ~1.8 eV in Ti L-edge and 2.5 eV in O K-edge spectra in all samples strongly matches the known spectra for the anatase phase and confirms that the oxidation state of Ti4+ has remained unchanged. Generally, owing to the localization of charge in Ti 3d orbitals in Ti3+, a shift in the absorption peaks to lower photon energy is observed. A change in the intensity of t2g and eg peaks with respect to dopant concentration is clearly seen. This change reveals whether or not the dopant atom is pulling the electrons away from Ti atom. As shown in Figure 3a, the sample with highest dopant content (ST3) shows the highest intensity, which is indicative of electrons leaving the Ti 3d orbitals due to the presence of neighboring Sr. Also, the relative intensities of the doublet in L3- eg, which is normally higher for the lower energy peak to the higher energy one in the anatase, is the same for all doped samples. A close inspection of the O K-edge spectra (Figure 3b) reveals slight changes in the intensity as well as the t2g- eg splitting with respect to doping content. The decrease in intensities of the doped samples is an evidence of a reduction in O 2p–Ti 3d bands, which is consistent with the higher intensity of the corresponding Ti L-edge NEXAFS.