Reports: DNI350843-DNI3: Trinuclear Copper Complexes for Dioxygen Activation and Reduction and Dinuclear Early Metal Catalysts for Olefin Polymerization
 printer friendly
printer friendlyMultimetallic enzymatic active sites perform diverse biological functions, including electron transfer, hydrolytic reactions, O2 reduction, water reduction and oxidation. In the context of O2 reduction to water, the tricopper active sites of laccase and ascorbate oxidase (AO) have been studied extensively given the potential applications in fuel cell chemistry. We have developed a novel strategy for the generation of trimetallic moieties based on templating a ligand scaffold around yttrium or lanthanides to generate a seven-coordinate complex that displays three multidentate sites for binding of transition metals. The previous progress report detailed mutimetallic O2 chemistry. The present report gives an update on the related tricopper chemistry and also describes a new direction in multimetallic catalysis, related to olefin polymerization.
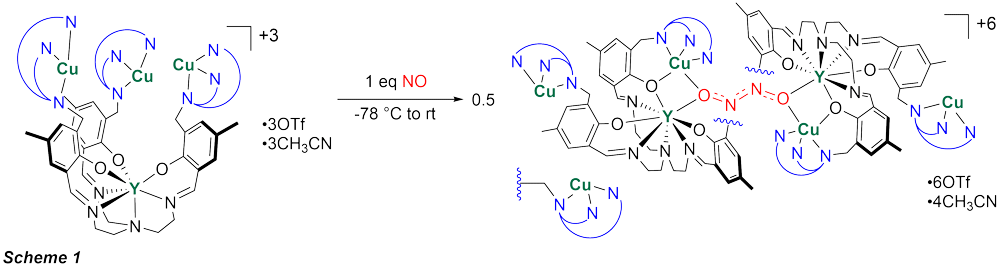
Reaction of previously prepared tricopper complex with ~1 equiv NO at low temperatures resulted in a new species that was crystallographically characterized (Scheme 1). The structure shows a dimer of the yttrium-tricopper complex bridged by a N2O2 moiety that is the result of reductive coupling of two NO molecules to form a N–N bond. At each Cu3Y monomer, the oxygen atom in the N2O22- motif bridges between the yttrium center and one of the pendant copper centers. The ligand scaffold, normally enveloping the yttrium center completely, has undergone a conformation change to allow access to yttrium and positioning of one of the copper centers in close proximity. As a consequence, the other two copper-bearing arms are pushed in the opposite direction. The copper centers not involved in binding to the hyponitrite moiety seem to have retained their +1 oxidation state and maintain their distorted tetragonal geometry (three nitrogen donors from the ligand plus one acetonitrile molecule). Therefore, as only one copper center is involved in reactivity on each Cu3Y monomer, the overall process can be described as a two-electron reductive coupling of two NO molecules. This type of activation of NO, involving two redox active and two redox inactive, Lewis acidic, metals is unique. This species provides precedent for a Lewis acid stabilized hyponitrite intermediate, at the site where protons bind in biological systems. Future studies will focus on the reactivity of the hyponitrite moiety, toward release of N2O.
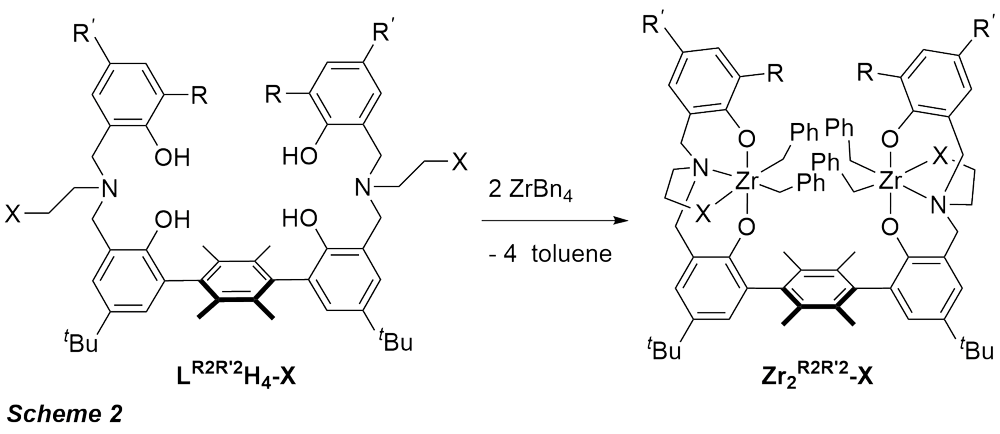
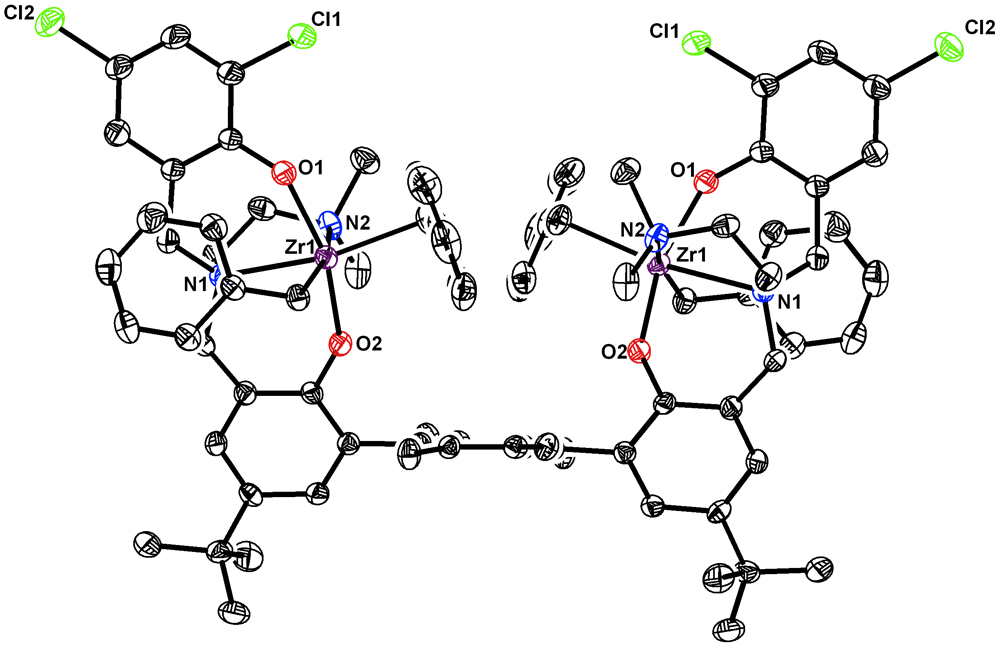
We have recently developed several dinuclear nickel catalysts for olefin polymerization. The bimetallic nature of these catalysts allowed for polymerization of ethylene in the presence of inhibitors such as amines, for the copolymerization of ethylene with aminoolefins. In order to expand the scope of bimetallic catalysts in olefin polymerization we designed ligands that can bind two early metals. Frameworks capable to support early metal-based dinuclear catalysts are of interest for higher activity, -olefin incorporation, and tacticity control. Permethylated terphenyl linker was employed to restrict rotation around aryl-aryl bonds and lock metal centers in close proximity. Di(amine bisphenolate) dizirconium systems analogous to CS and C1 symmetric monometallic systems reported by Kol and coworkers were prepared (Scheme 2). This ligand design was selected for its versatility, ease of synthesis, and interesting catalytic properties of monometallic Zr catalysts. Highly activeCS symmetric tetradentate amine bisphenolate zirconium complexes were initially reported by Kol et al., though these catalysts gave only atactic polymer. Only the syn atropisomers of these dinucleating ligand precursors (H4R2R'2-L) were synthesized, as the primary focus of these studies was to examine the effect of a proximal metal center. Spectroscopic and structural characterization supported the synthesis of the desired dizirconium complexes with the two metal centers in close proximity (Figure).
Monometallic and bimetallic catalysts were very active for 1-hexene polymerization causing a significant exotherm upon stoichiometric activation with [CPh3][B(C6F5)4]. Upon optimization of polymerization conditions, activities of up to 124,000 (g poly(1-hexene))(mmol Zr)-1(h)-1 were achieved for Zr2Cl4-NMe2. This is among the highest olefin polymerization activities reported for non-metallocene catalysts. At low temperatures (-30 °C), poly(1-hexene) with 79 % mmmm was obtained at 2,700 (g poly(1-hexene))(mmol Zr)-1(h)-1. This polymer also exhibited high molecular weight of 1.2×106 Da and low polydispersity (PDI=1.1). Under the same conditions, the best of the monometallic catalysts displayed 54 (g poly(1-hexene))(mmol Zr)-1(h)-1, and 33 % mmmm. Although the polymerization of propylene showed lower activity and isotacticity compared to 1-hexene, similar trends are observed. The most active catalyst is again Zr2Cl4-NMe2 with 47,000 (g polypropylene)(mmol Zr)-1(h)-1 and 43% mmmm. In contrast, a related monometallic catalyst showed significantly lower activity (2,900 (g polypropylene)(mmol Zr)-1(h)-1) and polymer tacticity (31% mmmm).
The present results indicate that the bimetallic catalysts display significantly enhanced activity and tacticity control compared to the monometallic analogs. The mechanism consistent with the reported results involves site epimerization following each insertion such that the polymer chain does not stay in between the two metal centers, a position which is disfavored by distal steric interactions. The increased isoselectivity of the bimetallic systems requires both local and distal steric control. On-going efforts are focused on expanding the applicability of the distal steric effect in bimetallic and monometallic catalysts toward the synthesis of stereoregular materials with other non-polar as well as functionalized monomers.
The above molecular architectures are expected to be useful for the assembly of multimetallic complexes for a variety of catalytic transformations, multielectron activation of small molecules to olefin and polar monomer polymerization. The ACS PRF funding has been instrumental starting (multicopper chemistry) and continuing (bimetallic polymerization) various aspects of this project. Two graduate students, and minority undergraduate have been funded on this grant. One of the graduate students is continuing his work on projects involving O2 chemistry, while the second is in the process of writing her PhD thesis; she will continue her career with a postdoctoral position in the area of synthesis of advanced new materials based on polyolefins. The undergraduate is currently a graduate student at Northwestern University.
Manuscripts (published or in progress):
Davide Lionetti, Michael W. Day, Theodor Agapie "Metal-Templated Ligand Architectures for Trinuclear Chemistry: Tricopper Complexes and Their O2 Reactivity" Chem. Sci., 2013, 4, 785-790
Madalyn R. Radlauer, Theodor Agapie "Bimetallic Zirconium Amine-Bisphenolate Polymerization Catalysts: Enhanced Activity and Tacticity Control for Polyolefin Synthesis" manuscript in preparation
Davide Lionetti, Theodor Agapie "Nitric oxide activation by multi-copper complexes" work in progress