Reports: DNI350840-DNI3: Bio-inspired Inorganic Catalysts for CO2 Reconversion
 printer friendly
printer friendlyIn the last funding period, we have developed novel deoxygenation of a mono-oxo bis(dithiolene) molybdenum and tungsten complexes, chemistry of which had been rarely studied due to the instability of the resulting deoxygenated products. We were able to achieve this chemistry through controlled protonation of the mono-oxo complexes.1 The significance of this work is two-fold: 1) this is the first example of the removal of oxide of mono-oxo bis(dithiolene) Mo/W complexes by protonaton, which mimics the chemistry of Mo/W containing oxotransferases. 2) The resulting deoxygenated products, [M(MeCN)2(S2C2Ph2)2], can serve in the future as a valuable synthon for model complexes for the enzyme active sites and the reaction intermediates.
Nature utilizes chemistry of high-valent metal oxo species to carry out a wide range of important catalytic reactions. Many heme and non-heme iron enzymes (e.g., cytochrome P450) use highly reactive FeIV=O for their oxygenase and oxidase activity. Photosynthesis relies on high-valent Mn=O species to carry out water splitting. In addition to Fe and Mn, nature also uses a metal-oxo motif with a second and a third row transition metal ions such as Mo and W which have considerably different chemical properties from those of Fe- and Mn-oxo species. In contrast to the highly reactive Fe/Mn oxo counterparts, molybdenum and tungsten in high oxidation state (IV-VI) are stabilized by the oxo ligand. The high valent Mo=O or W=O species are often considered a thermodynamic sink in synthesis because they can be easily generated by a trace amount of O2 or water contamination. Nature, however, efficiently utilizes the Mo=O or W=O unit to achieve difficult multi-electron redox catalysis of oxotransferases, in which the addition and removal of the oxo group of the Mo/W center is elegantly coordinated as involving water, protons, and electrons.
One of the best studied oxotransferases is dimethylsulfoxide reductase (DMSOR) from R. sphaeroides. DMSOR catalyzes the reduction of dimethylsulfoxide (DMSO) to dimethylsulfide (DMS). The X-ray structure reveals that the active site in the reduced state has a penta-coordinate MoIV ion ligated by two pterin-dithiolene cofactors and a serine side chain, while the oxidized state has an additional oxo group at the MoVI center (Scheme 1a). The catalytic cycle begins with an oxygen atom transfer from DMSO to the MoIV center in the oxidative half-cycle (DMSO + MoIV ¨ DMS + MoVI=O). In the reductive half-cycle, the MoIV site is restored by removing the oxo group assisted by proton/electron transfer (MoVI=O + 2 e– + 2 H+ ¨ MoIV + H2O). The O-atom transfer chemistry in the oxidative half-reaction has been extensively studied with synthetic bis(dithiolene) metal (Mo or W) complexes by a group of researchers (Scheme 1b). The comparative studies between Mo and W complexes with various substrates by Holm and coworkers showed that the ligand electronic environment as well as the metal ion can affect the oxo-transfer reaction rate, XO + MIV ¨ X + MVI=O, where M = Mo, W; X = sulfides, amines, and others (Scheme 1b).
Scheme 1
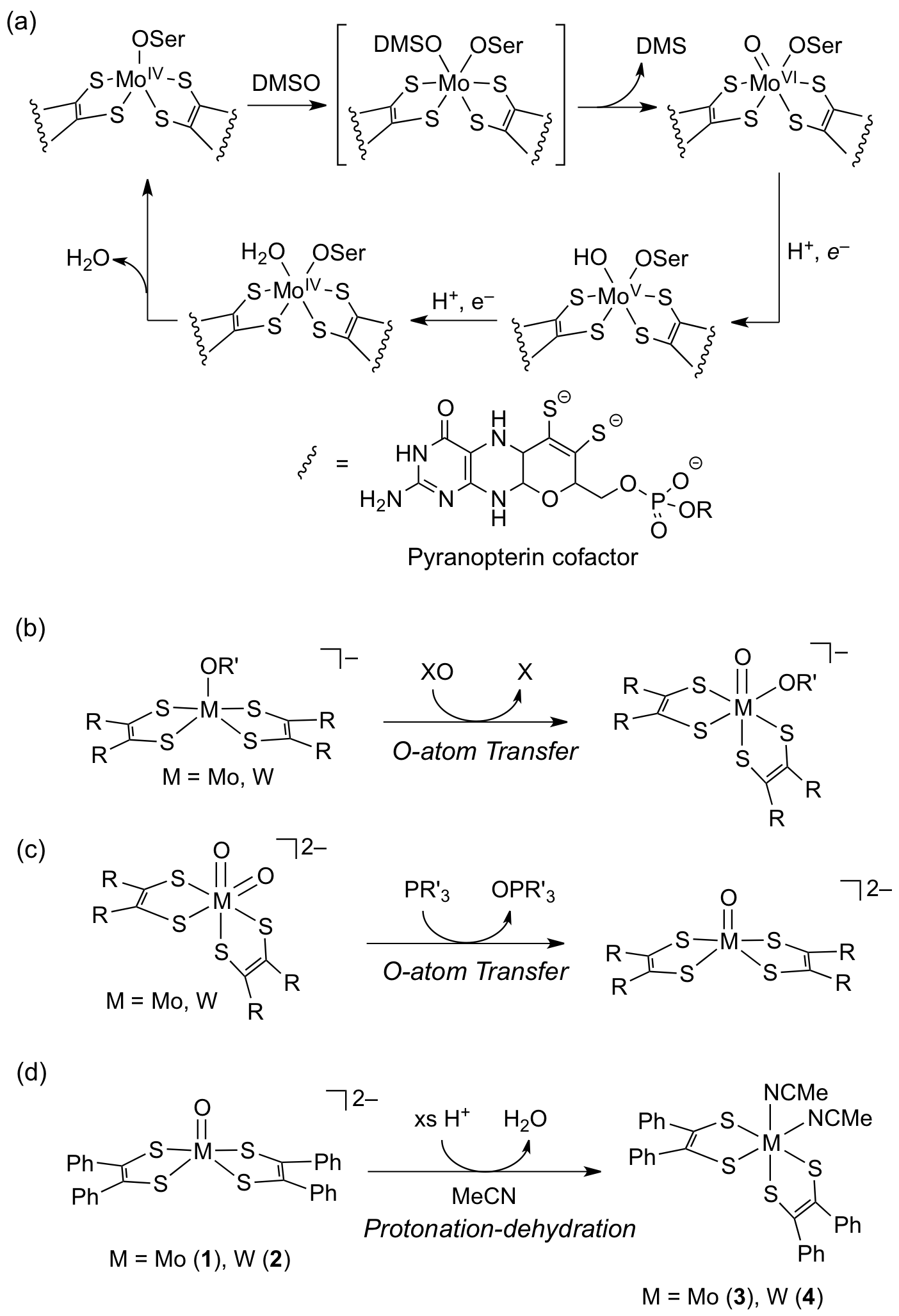
In contrast to the oxidative half-reaction, it had been a challenge to achieve deoxygenation chemistry with this group of complexes because the removal of the oxo ligand often causes a complex decomposition to tris(dithiolene) Mo/W species. The only successful way of removing the oxo group from the bis(dithiolene) metal site appears to be the use of non-biological oxygen abstractors like phosphines (e.g., PPh3) by which the O-atom, as opposed to oxide (O2–), can be removed from the metal site with a concomitant reduction of the metal ion (Scheme 1c).
Inspired by the biological process of converting a metal-bound oxo ligand to water, we have studied protonation chemistry of discrete molybdenum and tungsten oxo complexes. Our study1 demonstrates that simple protonation can efficiently remove the oxo group from the mono-oxo bis(dithiolene) Mo or W complexes and the resulting deoxygenated products can be stabilized by solvent. Protonation of [MoIVO(S2C2Ph2)2]2– (1) and [WIVO(S2C2Ph2)2]2– (2) in acetonitrile efficiently generates novel deoxygenated complexes, [Mo(MeCN)2(S2C2Ph2)2] (3) and [W(MeCN)2(S2C2Ph2)2] (4), in which the coordinated MeCN ligands can be easily substituted by a stronger ligand such as CO and PPh3. While CO replaces both MeCN ligands to yield dicarbonyl products, [M(CO)2(S2C2Ph2)2], a sterically demanding incoming ligand like PPh3 results in a mono-substitution, [Mo(MeCN)(PPh3)(S2C2Ph2)2] (7). Differential reactivity between [MoIVO(S2C2Ph2)2]2– (1) and [WIVO(S2C2Ph2)2]2– (2) with protons has been observed. Whereas 3 is the sole product from the reaction between 1 and acid, two different reaction products, [WO(S2C2Ph2)2]– and [W(MeCN)2(S2C2Ph2)2] (4), can be generated from 2 depending on the amount of acid added. The former is predominantly generated with one equivalent of acid while the latter is the main product with an excess amount of acid.
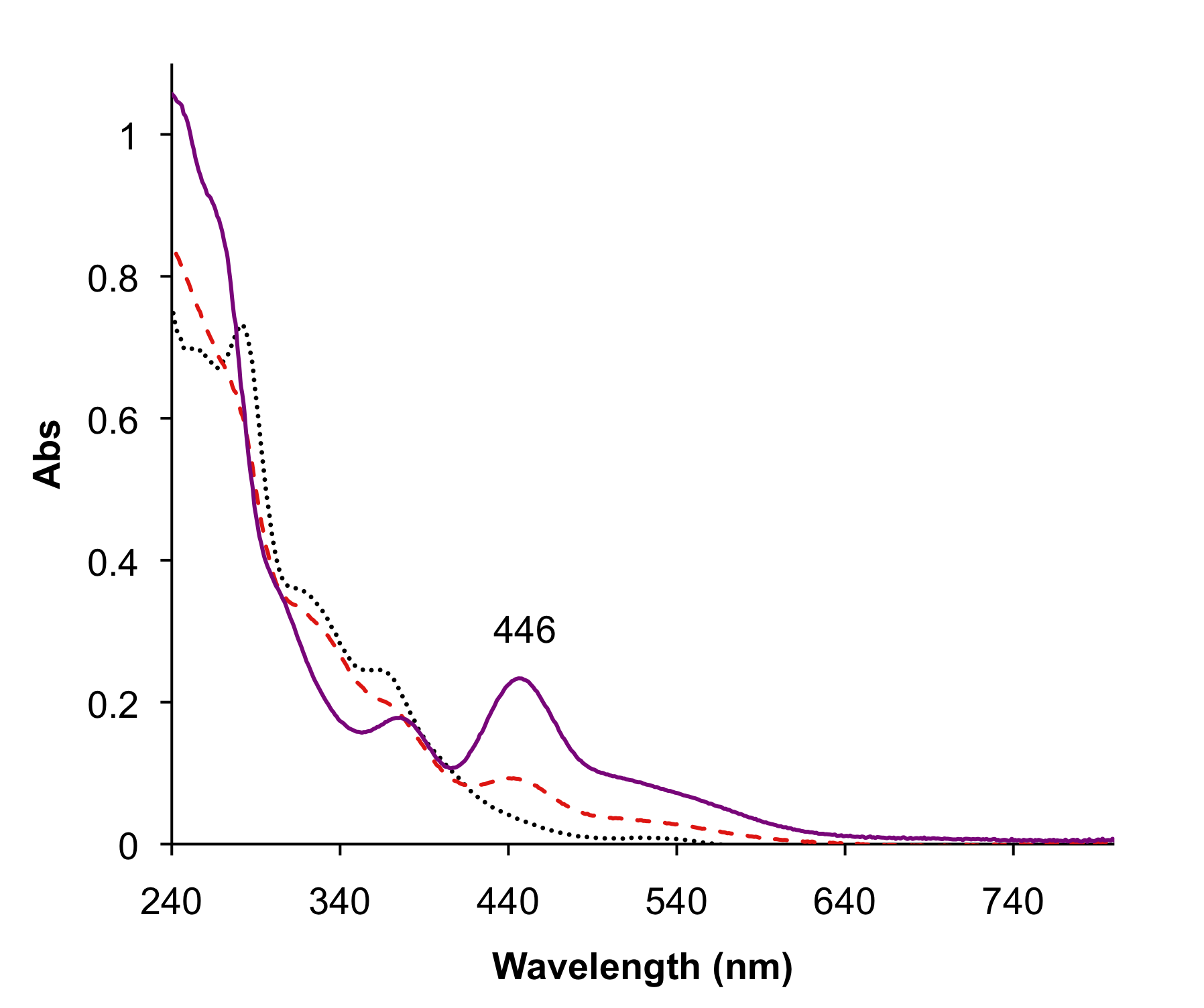
Figure 1. UV-Vis spectral changes during titration of (Et4N)2[MoO(S2C2Ph2)2] (1) (black dotted) with tosylic acid (TsOH) in acetonitrile at room temperature. An addition of 1 equiv of TsOH to 1 results in the incomplete conversion (red dashed) of 1 to [Mo(MeCN)2(S2C2Ph2)2] (3). A further addition of extra 1 equiv of TsOH leads to the complete formation of 3 (purple solid).
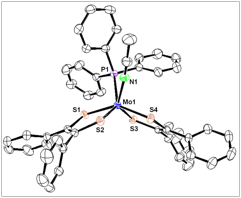
Figure 2. Structure (ORTEP view) of Mo(PPh3)(MeCN)(S2C2Ph2)2 (7) with 50 % probability thermal ellipsoids. Hydrogen atoms were omitted for clarity.
(1) Seo, J.; Williard, P. G.; Kim, E. Inorg. Chem. 2013, 52, 8706-8712









