Reports: DNI352452-DNI3: A Nonmetal Platform for Inner Sphere Two-Electron Redox Catalysis
 printer friendly
printer friendlyThis ACS PRF grant supports investigations into the catalytic hydrogen transfer chemistry of a phosphorus-based platform operating in a P(III)/P(V) redox couple. We have previously shown[1] that planar T-shaped σ3-phosphorus compound 1[2] dehydrogenates ammonia-borane (H3N•BH3) to give pentacoordinate dihydridophosphorane 1•[H]2, and that 1•[H]2 in turn is capable of transferring hydrogen to azobenzene to give 1,2-diphenylhydrazine and regenerate 1 (Figure 1). Consistent with this stoichiometric reactivity, we have demonstrated that both phosphorus species 1 and 1•[H]2 promote the transfer hydrogenation of azobenzene with NH3BH3 in a catalytic fashion.
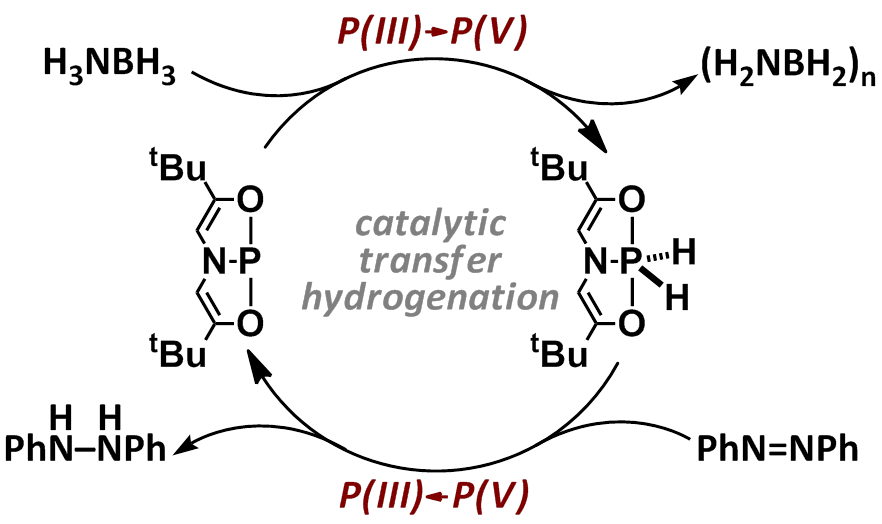
Figure 1. Catalytic hydrogen transfer reactivity of 1/1•[H]2.
One of the central goals of the PRF-funded research was to define the fundamental reactivity permitting this catalytic oxidation state and coordination number cycling at phosphorus. To this end, we have undertaken mechanistic studies of the hydrogen transfer reaction from 1•[H]2 to azobenzene (Figure 2). The course of the reaction can be followed by monitoring consumption of 1•[H]2 with respect to time by either 1H or 31P NMR spectroscopy. With a 10-fold excess of azobenzene acceptor, the hydrogen transfer reaction from 1•[H]2 obeys pseudo-first order kinetics. The observed rate constant kobs is found to depend linearly on the concentration of azobenzene over the range 0.03-1.58 M, consistent with an overall second order rate law given as: v = k [1•[H]2]•[PhN=NPh]. Activation parameters (ΔH╪ = 12.4±0.7 kcal/mol, ΔS╪ = –36±6 cal/mol•K) derived from Eyring analysis over the temperature range 10–40 °C are unremarkable for a bimolecular reaction of this type. Treatment of 1•[H]2 with N-deuterated primary amines (i.e. BnND2) results in H/D exchange at phosphorus, permitting the preparation of isotopologue 1•[D]2. The reduction of azobenzene with 1•[D]2 gives kH/kD = 3.76±0.46, indicative of the transferal of at least one hydrogen atom in the rate determining step.

Figure 2. Kinetic investigation of hydrogen transfer from 1•[H]2 to azobenzene.
A Hammett study of symmetrically para-disubstituted azobenzenes shows a linear free energy relationship between the rate of reduction and increasing electron withdrawing character of the substituent. The Hammett sensitivity constant (ρ=2.4) is consistent with the accrual of negative charge on the azo acceptor during the rate limiting step, as would be expected in a hydride transfer mechanism.
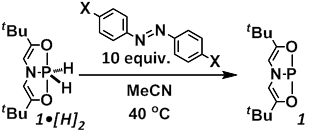
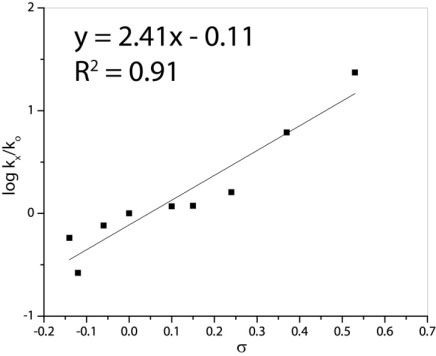
Figure 3. Hammett correlation for hydrogen transfer from 1•[H]2 to p-substituted azobenzenes. ρ=2.4, R2=0.90.
Consistent with the hydricity implied by the Hammett study, compound of 1•[H]2 undergoes hydride abstraction with trityl tetrafluoroborate (Ph3CBF4). Investigations are underway to quantify the hydricity of 1•[H]2 in a more rigorous fashion.
In summary, support from the ACS-PRF during the first year of the project has funded the research of one graduate student who has made substantial progress toward understanding the fundamental reactivity of 1•[H]2. The next grant year will be spent focusing on broadening the scope of the catalytic transfer hydrogenation reactivity of 1/1•[H]2 with respect to substrate and hydrogen donor.
[1] Dunn, N.L.; Ha, M.; Radosevich, A.T. J. Am. Chem. Soc., 2012, 134, 11330.
[2] a) Culley, S.A.; Arduengo, A.J. J. Am. Chem. Soc. 1984, 106, 1164. b) Arduengo, A.J.; Stewart, C.A.; Davidson, F.; Dixon, D.A.; Becker, J.Y.; Culley, S.A.; Mizen, M.B. J. Am. Chem. Soc. 1987, 109, 627. c) Arduengo, A.J.; Stewart, C.A. Chem. Rev. 1994, 94, 1215.









