Reports: DNI351472-DNI3: Electrocatalysts for the Proton-Coupled Electron Transfer Promoted Reduction of Carbon Dioxide
 printer friendly
printer friendlyMost previously studied CO2 reduction catalysts display poor selectivities, limited stabilities and require large overpotentials to effect CO2 activation at an appreciable rate. These shortcomings can be overcome by judicious ligand and catalyst design. For example, the instability of some of the most successful CO2 reduction catalysts is due to the high lability of the ligand scaffolds used to support low-valent catalytic centers. The sluggish kinetics observed for such platforms can be attributed to the fact that these systems do not minimize the large nuclear reorganization energy associated with binding of CO2 to reduced metal centers. In confronting these issues, we have developed a new kinetically robust platform for CO2 conversion and are designing platforms with hydrogen-bonding functionalities to stabilize M–CO2– adducts and promote the controlled delivery of protons to drive C–O bond cleavage to efficiently generate CO and H2O. In pursuing these research opportunities, we have identified the following objectives.
The development of transition metal carbene complexes for CO2 activation has been a major area of focus in our lab.[1] In carrying out this work, we have developed complexes of Ni and Pd supported by the Pyridyl-DiCarbene (PDCR) ligand scaffolds depicted in Figure 1A. The two NHC moieties on the ligand backbone support the electron-rich Ni and Pd metal centers needed for catalysis and are suitable for activating CO2. Moreover, the modular synthesis of these compounds allows for the steric and electronic properties of these architectures to be tuned with ease and fidelity. Crystal structures for a homologous set of these palladium CNC-pincer complexes are shown in Figure 1B. Inspection of these ORTEP diagrams clearly illustrates how the size and accessibility of the molecular cleft is attenuated by variation of the pincer R-group. Attenuation of the steric bulk about the pincer periphery significantly impacts the kinetics and efficiacy and catalysis at the metal center.
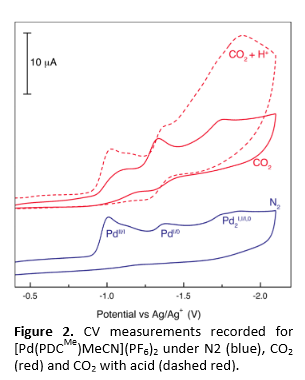
We have successfully shown that the palladium systems of Figure 1A are competent electrocatalysts for reduction of CO2. Shown in Figure 2 are the cyclic voltammetry traces recorded for [Pd(PDCMe)MeCN](PF6)2 in MeCN. The CV trace recorded under N2 (Figure 2, blue) reveals the redox activity of the Pd-pincer architecture with reduction waves centered at E° ~ –1.0 V and –1.3 V (vs. Ag/AgCl), which correspond to PdII/I and PdI/0 couples, respectively. The pincer constructs of Figure 1 proficiently catalyze the electrochemical conversion of CO2 to CO. Bulk electrolysis of solutions of these complexes in acidic MeCN under an atmosphere of CO2 has allowed for the products of the electrocatalytic reduction to be ascertained by gas chromatography. Our Pd(PDC) architectures display exquisite selectivity for CO production over other reduced carbon species including CH3OH, HCO2H and H2CO. Moreover, we have shown that H2 is not formed as an unwanted side product via direct reduction of protons by the Pd catalyst. Accordingly, our novel Pd(PDC) construct displays far higher stability, selectivity and efficiency compared to homologous Pd-phosphine complexes.
A number of heterogeneous cathode materials can also facilitate the electrocatalytic conversion of CO2 to CO, however only noble metals such as Ag and Au can catalyze this reaction with Faradaic Efficiencies (FEs) that are in excess of 80% at ambient pressures.[2] The production of CO using precious metal cathodes has been hampered by the exorbitant cost of these materials, which eliminates their practical use on the scale required for alternative fuel synthesis. Against this backdrop, our group recently developed a bismuth-based Carbon Monoxide Evolving Catalyst (Bi-CMEC), which can convert CO2to CO at an appreciable rate (high current density; jCO) with a FE over 90% at small overpotential (h = 165 mV).[3]
We have shown that Bi-CMEC can be conveniently electrodeposited onto a glassy carbon electrode (GCE) by controlled potential electrolysis (CPE) of a quiescent solution of Bi3+ in aqueous acid.3 SEM revealed that this Bi containing material is composed of an array of striated clusters (Figure 3). Cyclic voltammetry (CV) experiments showed that the Bi material could electrocatalyze reduction of CO2 in MeCN, and that the kinetics and efficiency of this process were greatly enhanced by addition of small amounts of imidazolium based ionic liquids (ILs) such as 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM]BF4) to the catholyte solution (Figure 4).3 Conversion of CO2 to CO using Bi-based cathodes represents an important development in the fields of CO2 electrocatalysis and renewable energy conversion. Our development of Bi-CMEC and its significance to these fields was recently highlighted in a feature article in Chemical and Engineering News.[4] As such this PRF award has had a significant effect on my career by allowing me to establish a research program that is making fundamental discoveries in the areas of CO2 catalysis and renewable energy conversion. This award has also positively affected each of the students in my research lab (both graduate and undergraduate) by providing support and the means to develop our expertise in these areas.



References:
[1]. Ariyananda, P. W. G.; Yap, G. P. A.; Rosenthal, J. “submitted.
[2]. Hori, Y., Wakebe, H., Tsukamoto, T., Koga, O. Electrochimica Acta 1994, 39, 1833–1839.
[3]. DiMeglio, J. L.; Rosenthal, J. “J. Am. Chem. Soc. 135, 8798–8801.
[4]. Jacoby, M. Chem. Eng. News, 2013, 91, 21–22.









