Reports: UNI150337-UNI1: Development of a Facile Tandem Asymmetric Epoxidation/Ring-Opening Protocol: An Efficient Entry to Enantiomerically Enriched Tertiary Naphythoquinols
 printer friendly
printer friendlyAs stated in my project proposal, the goals of my research were: (1) to develop a highly efficient method for the construction of either enantiomer of chiral naphthoquinols via a tandem oxidation/ring-opening of cyclic 3,4-epoxyalcohols; (2) to employ the proposed methodology in conjunction with a tandem intramolecular biaryl ether formation/phenolic oxidation sequence to access the core framework of the natural products spiroxins A-E in an asymmetric fashion. Towards these goals, I have trained ten students who have worked on the development and optimization of individual steps of the proposed research. In the past two years, we have successfully completed the first goal of the project, finalized all the data collection for this goal and published a manuscript in Tetrahedron Letters.1 In addition, we began work towards the second goal of the project and currently we have well-established scale up procedures for several steps of our synthetic protocol to prepare enantioenriched tertiary naphthoquinols en route to the synthesis of the spiroxins core structure.
In 1999, the natural product spiroxin A was isolated from a marine fungus and was found to exhibit promising antitumor activity against human ovarian carcinoma in mouse xenografts. Since the discovery of spiroxin A, its biological properties have been little explored despite its promising bioactivity against ovarian cancer cells. This discrepancy may be explained by spiroxin A's limited availability, either through total synthesis or by isolation from natural sources. Spiroxin A has proven synthetically challenging due to its condensed structure, which contains six stereogenic centers within a twenty-carbon framework. The important biological properties of spiroxin A, and its fascinating molecular structure, prompted my research lab to design a novel synthetic approach towards this natural product. Our approach towards its total synthesis proceeds through a key chiral tertiary naphthoquinol intermediate followed by the construction of the octacyclic core.
The key tertiary naphthoquinol, besides being an important synthetic intermediate en route to spiroxin A, is also present in other natural products with important biological activities. In spite of this fact, almost no methods exist for the asymmetric preparation of quinols from achiral precursors. Therefore, in the past two years, our laboratory developed a novel asymmetric synthesis of chiral tertiary naphthoquinols (Goal 1, Figure 1). In addition to completing all the proposed synthetic steps, we have also unambiguously characterized all of the synthetic intermediates and performed optimization studies to prepare several of the synthetic intermediates.
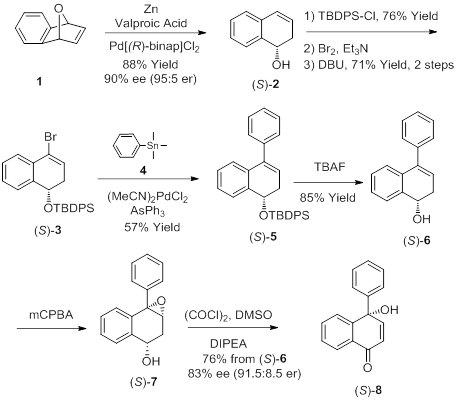
Figure 1. General asymmetric method to access chiral tertiary naphthoquinols
Goal 2 of the proposed project consists of employing the new methodology developed by our laboratory (Goal 1) in conjunction with a tandem intramolecular biaryl ether formation/phenolic oxidation sequence to access the core framework of the spiroxins. As the first step towards this goal, the applicability of our novel method to compounds of increasing structural complexity must be evaluated. Over the past year, we have been investigating the versatility of our asymmetric method by synthesizing three analogues of the (S)-8 chiral tertiary naphthoquinol (Figures 1 and 2).
To prepare these new chiral naphthoquinols in the highest possible overall yield, optimization studies for the first few steps have been performed as these steps are common to the various analogues. To this end, a large-scale stereoselective ring opening of cyclic ether 1 to prepare the homoallylic alcohol (S)-2 was tested. Several reaction parameters studies, including catalyst source, extraction conditions, and chromatographic conditions were thoroughly investigated. As a result of these efforts, we are now able to scale up this reaction step with consistently high yields. Considerable work was also placed on optimization of the second step which consists of silylation of homoallylic alcohol (S)-2 to generate the TBDPS-protected (S)-2 silyl ether. Chromatographic conditions were optimized and scale-up reaction conditions were successfully identified for this step. In an effort to reduce the number of purification steps in the overall synthetic sequence, we also tested the efficacy of the silylation reaction using crude homoallylic alcohol (S)-2. Our early studies revealed the vinyl bromide derivative (S)-3 to be an unstable intermediate with a short shelf-life, even when held at low temperature. Hence, it must be used in the next step soon after it has been prepared. During this year, we have performed studies to determine the stability window of this compound and have successfully identified optimal storage conditions to prolong its stability. With these optimization studies complete, we are now in an excellent position to complete the synthesis of derivatives (S)-9, (S)-10 and (S)-11 (Figure 2).
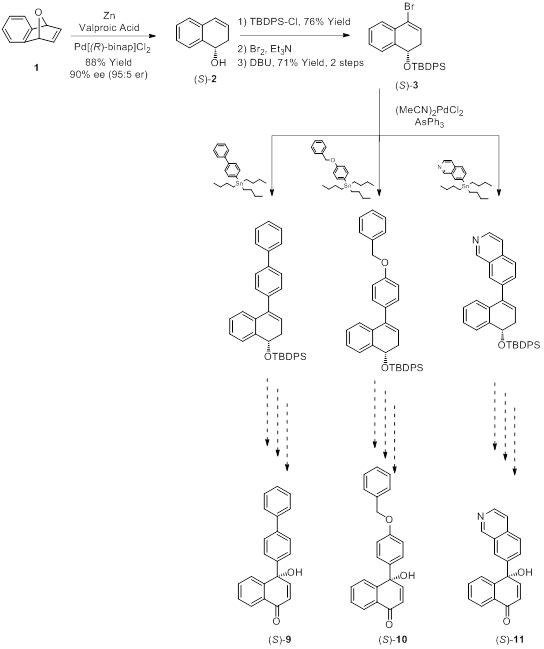
Figure 2. Testing versatility of asymmetric method
The results of these research efforts were the basis for presentations at the 2011 Wellesley College Ruhlman Conference, the 241st ACS National Meeting and the 2013 Wellesley College Summer Research Program symposium, as well as a Tetrahedron Letters manuscript. They will also be presented at the 247th ACS National Meeting and the 2014 Wellesley College Ruhlman Conference.
Through the generous support of the Petroleum Research Fund, I was able to train several students in my laboratory over the past few years. The research opportunities I was able to provide in my lab with the help of this award gave my students the opportunity to conduct scientific research and to develop a real appreciation for the importance of basic research in chemical education to understand and explain the world around us. It gave them the opportunity to apply what they have seen in their organic chemistry classes in systems that they have never seen before and extend their understanding of the organic chemistry field.
For the upcoming year, we will continue our work towards testing the versatility of our methodology en route towards the total synthesis of the core framework of the spiroxins and will also begin to evaluate the cytotoxicity of individual synthetic intermediates against ovarian cancer cells. These studies will be invaluable for continuing research towards our long term goal of accomplishing the enantioselective total synthesis of spiroxin A and conducting structure activity relationship (SAR) studies of spiroxin A analogues.
References:
1. Kwan, A.; Stein, J.; Carrico-Moniz, D.* Tetrahedron Lett. 2011, 52, 3426-3428.









