Reports: UR151961-UR1: Development of a Catalytic, Asymmetric Aza-Cope Rearrangement and Mannich Cyclization
 printer friendly
printer friendlyScheme 1
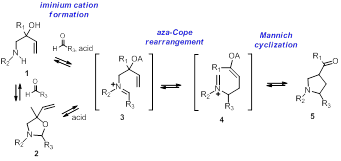
We have recently found that acyl pyrrolidine 5a can be accessed rapidly with high trans diastereoselectivity by treating a ca. 2:1 mixture of oxazolidine diastereomers 2a with 10 mol% BF3OEt2 for 15 minutes at room temperature (Scheme 2) [3,4]. To determine the scope and limitations of this selectivity, we have synthesized oxazolidines 2b, 2c, and 2d, and are in the process of subjecting them to aza-Cope—Mannich conditions. Interestingly, a 1:1 mixture of diastereomeric oxazolidines 2b required more forcing conditions (50 mol% BF3OEt2, 3.5 hours at 35 oC) to achieve ca. 75% conversion to a 6:1 mixture favoring trans formal pyrrolidine 5b. Likewise, we have thus far been unable to observe any reaction whatsoever when oxazolidine 2d is exposed to as much as 50 mol% BF3OEt2 and stirred for up to 24 hours. We are in the process of exploring higher temperature reaction conditions to achieve some conversion. We have very recently synthesized oxazolidine 2c, albeit in very low yield. We anticipate that the results of the reaction of this oxazolidine will shed some light on the relative effects of R1 and R2 on the rate of the aza-Cope—Mannich reaction.
Scheme 2
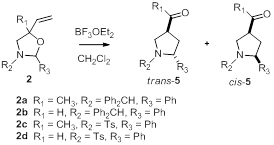
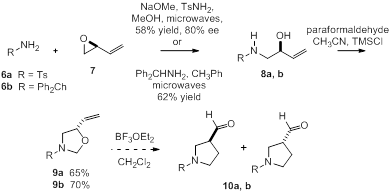
The above examples address diastereoselectivity issues in the aza-Cope—Mannich reaction, but do not probe enantioselectivity since the oxazolidine reactants described are all racemic. Prior to the beginning of this award, synthesized oxazolidines 6a and 6b as single enantiomers and subjected those to our aza-Cope—Mannich conditions (Scheme 3). We were delighted to find that oxazolidine 6a formed acetyl pyrrolidine 7a in good yield and diastereoselectivity. However, oxazolidine 6b reacted to form formyl pyrrolidine 7b with much poorer selectivity, presumable due to the conformational mobility of the substrate.
Scheme 3
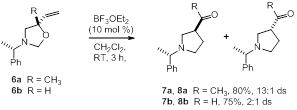
However, when a 50:50 mixture of oxazolidine 6a and its carbinol diastereomer 8 were subjected to the same conditions, the diastereoselectivity fell to 1.3:1 (Scheme 4), suggesting that the chiral auxiliary has a minimal impact on the stereochemical outcome. It indicates that asymmetry is likely induced by memory of chirality [5] rather than by the influence of the chiral amine protecting group. If this is the case, this is the first example of such asymmetric induction in a conformationally mobile system.
Scheme 4
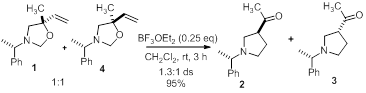
To further probe this issue, we have synthesized enantioenriched oxazolidines 9a and 9b via microwave-assisted epoxide aminolysis of commercially available S-butadiene monoxide 7 [6]. Amino alcohol 8a was accessed in 58% yield but only 80% ee according to chiral HPLC analysis. We are currently in the process of determining the ee of amino alcohol 8b. These oxazolidines 9a and 9b will then be subjected to aza-Cope—Mannich conditions to determine whether the enantioselectivity may be achieved from substrate control in the absence of a chiral auxiliary.
Scheme 5
Several undergraduate students have participated in this project during this reporting cycle. Four of those students are still undergraduates at Eastern Michigan University; the majority of them plan to attend graduate school in chemistry in the next one to three years. Two students who graduated last spring are now enrolled in graduate school at Indiana University and Scripps Research Institute, FL. An ACS Project SEED student who worked on this project for two years is now a freshman at Yale University. Last year four students presented posters at national American Chemical Society meetings; this year two more students will present posters at the ACS national meeting.
References
[1] Overman, L. E.; Kakimoto, M.-A. J. Am. Chem. Soc. 1979, 101, 1310-1312.
[2] For reviews of the aza-Cope rearrangement—Mannich cyclization and related reactions, see (a) The Intramolecular Mannich and Related Reactions Overman, L.; Ricca, D. in Comprehensive Organic Synthesis, Trost, B. M., Ed.; Pergamon: New York, 1991; Vol. 8, pp 1007-1046; (b) Overman, L. Acc. Chem. Res. 1992, 25, 352-359; (c) Overman, L. E. Aldrichimica Acta 1995, 28, 107-120; (d) Bonin, M.; Micouin, L. Chem. Rev. 2004, 104, 2311-2352; (e) Overman, L. E. Tetrahedron 2009, 65, 6432–6446.
[3] For FeCl3/TMSCl mediated aza-Cope—Mannich reaction, see Carballo, R. M.; Purino, M.; Ramirez, M. A.; Martin, V. S.; Padron, J. I. Org. Lett. 2010, 12, 5334.
[4] For BF3OEt2 mediated iminium cation formations in the aza-Cope-Mannich reaction, see (a) Overman, L. E.; Shim, J. J. Org. Chem. 1991, 56, 5005; (b) Overman, L. E.; Shim, J. J. Org. Chem. 1993, 58, 4662.
[5] Zhao, H.; Hsu, D. C.; Carlier, P. R. Synthesis 2005, 1. <pcxsplast">[6] Desai, H.; D'Souza, B. R.; Foether, D.; Johnson, B. F.; Lindsay, H. A. Synthesis 2007, 902.









