Reports: DNI152233-DNI1: A New, Reductive-Heck Approach towards Carbon(sp2)-Carbon(sp3) Bond Formation
 printer friendly
printer friendlyThe efficient construction of Csp2-Csp3 bonds remains a serious challenge in synthetic organic chemistry. Although remarkable advances have been made recently, these methods are limited by stoichiometric use of main-group organometallics, excess activating agents, poor atom economy and limited substrate scope. For example, the pre-functionalization of one coupling partner as a main-group organometallic nucleophile leads to significant synthetic inefficiencies and waste streams. These problems can be addressed by using simple, readily available olefins and alkynes in sp3-carbon construction. The use of olefins as cross-coupling partners in Csp2-Csp3 coupling requires interrupting the catalytic cycle of the Heck reaction before b-hydride elimination occurs. The work supported by the donors of the ACS-PRF seeks to expand the utility of the reductive-Heck reaction to include a range of readily available substrates currently incompatible with this process. During the first year of funding, several important observations have been made: 1) alkyl-palladium reduction has been shown to outcompete b-hydride elimination in a reductive-Heck reaction; 2) a formal synthesis of englerin A using the proposed reductive-Heck technology; 3) alkyne insertion has been demonstrated to outcompete b-hydride elimination; and 4) the alkyne insertion has been used in the synthesis of tri- and tetra-substituted olefins.
During a recent campaign to establish a versatile synthesis of the englerin family of natural products, we implemented a new approach to Csp2-Csp3 cross-coupling reactions by interrupting the Heck reaction with formate reduction prior to b-hydride elimination (Scheme 1).[1] This reductive-Heck reaction abrogates the need for main-group organometallics typically used in Csp2-Csp3 cross-coupling reactions. Consequently, the reaction holds promise as a general cross-coupling reaction due to the vast availability of olefins and enhanced step economy. In the context of natural product synthesis, the reaction provides a new approach to the hydroazulene ring system, a privileged structure in biologically active natural products, and allows stereochemical control over the critical ring junction. Furthermore, additional chemistry uncovered during the course of our englerin studies provided over 30 new natural product analogs that provided two active lead molecules for diabetes and tuberculosis (TB) in Lilly's PD2 screening program.

As a result of our discovery that formate reduction can outcompete b-hydride elimination, we have focused on identifying and understanding alternative mechanistic pathways to b-hydride elimination in Csp3-carbopalladium intermediates. Specifically, we utilize Csp3-carbopalladium intermediates generated from oxidative addition of alkyl electrophiles in unique migratory insertion pathways (e.g. Scheme 2). We described the first example of alkyne migratory insertion into Csp3-carbopalladium intermediates that provides a unique approach to the synthesis of tetrasubstituted olefins (Scheme 2 right).[2] During the course of this work, we have demonstrated that migratory insertion of alkynes into Csp3–palladium species can out-compete β-hydride elimination without exotic ligands. This process allowed the development of a tandem alkyne insertion/Suzuki reaction of unactivated alkyl iodides. The reaction has broad substrate scope: applicable to both primary and secondary alkyl iodides and useful in the construction of both five-membered and six-membered rings. Cursory mechanistic studies have led to the proposal of a plausible catalytic cycle. The overall transformation provides rapid access to regio- and stereodefined, unsymmetrical, tetrasubstituted olefins through a mechanistically unique reaction manifold which complements existing methods.

An extension of this work replaces the main-group organometallic with a hydride source to provide trisubstituted olefins (Scheme 2 left).[3] The new route to trisubstituted olefins proceeds through a palladium-catalyzed alkyne insertion/reduction reaction with unactivated alkyl iodides. The reaction occurs under mild conditions and tolerates a range of functional groups and substitution patterns. Preliminary mechanistic inquiry suggests that the transformation may proceed through a hybrid radical/organometallic pathway.
The move to secondary iodides has led to the discovery of an efficient, palladium-catalyzed iodine-transfer reaction of secondary alkyl iodides (Scheme 3).[4] Under the reaction conditions, an intramolecular double insertion of an alkyne and olefin provides access to primary iodides possessing β-hydrogens. The process delivers these complex bicyclic homoallylic iodides with stereo- and regio-defined tetrasubstituted olefins from easy-to-access secondary iodides. The reaction tolerates a variety of functional groups, including common heterocycles. Mechanistic studies suggest a role for palladium beyond simple radical initiation.
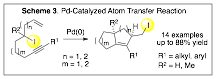
During the course of this research, we found that internal alkynes aid in the oxidative addition of primary iodides by palladium(0). This observation has led to the investigation of alkynes for palladium catalysis. Through continued efforts in this area, we will systematically define the requirements for fundamental organometallic processes capable of outcompeting b-hydride elimination in alkylpalladium species. This information will be valuable for the design of new palladium-mediated reaction manifolds that allow complexity-building cross-coupling reactions.
[1] Gao, P.; Cook, S. P. "A Reductive-Heck Approach to the Hydroazulene Ring System: A Formal Synthesis of the Englerins," Org. Lett. 2012, 14, 3340–3343.
[2] Monks, B. M.; Cook, S.P. "Palladium-Catalyzed Alkyne Insertion/Suzuki Reaction of Alkyl Iodides" J. Am. Chem. Soc, 2012, 134, 15297–15300.
[3] Fruchey, E. R.; Monks, B. M.; Patterson, A. M.; Cook, S. P. "Palladium-Catalyzed Alkyne Insertion/Reduction Route to Trisubstituted Olefins" Org. Lett. 2013, 15, 4362–4365.
[4] Monks, B. M.; Cook, S.P. "Palladium-Catalyzed, Intramolecular Iodine-Transfer Reactions in the Presence of b-Hydrogens" Angew. Chem. Int. Ed., 2013, accepted.









