Reports: ND350971-ND3: Electrocatalytic Water Oxidation by Manganese Pyridinophane Complexes
 printer friendly
printer friendlyWe previously reported that certain manganese pyridinophane complexes are hydrogen peroxide disproportionation catalysts (i.e. catalase mimics). We undertook a detailed investigation into the mechanism of hydrogen peroxide disproportionation by high spin [(Py2NMe2)Mn]2+ (Fig. 1, R = Me) which is active in aqueous solution over a wide pH range. This molecular catalase mimic is unusually robust and long-lived, achieving turnover numbers of almost 60000.
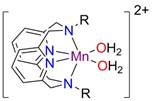
Fig. 1. [(Py2NR2)Mn]2+ complexes.
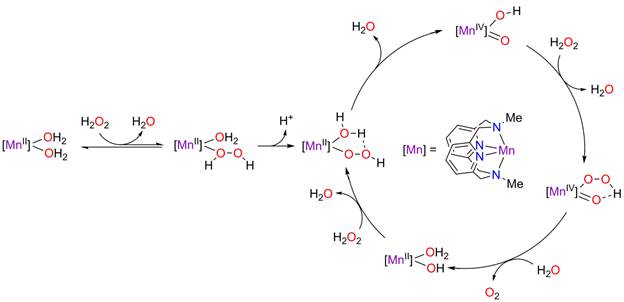
Initial rates measurements over a range of temperatures combined with kinetic isotope measurements, spectroscopic measurements under turnover conditions and a series of control experiments to eliminate the possibility of hydroxyl radial or superoxide formation led us to propose a unique mechanism for H2O2 disproportionation (Scheme 1).
Entry into the catalytic cycle involves substitution of an aqua ligand by H2O2, followed deprotonation to yield the Mn(II) aqua/hydroperoxo complex [Mn(Py2N2)(H2O)(O2H)]2+. Slow formation of [Mn(Py2N2)(H2O)(O2H)]+ accounts for the observed induction period. Aqua ligand-assisted intramolecular O-O bond heterolysis in the hydroperoxo complex provides the Mn(IV) oxo/hydroxo intermediate [Mn(Py2N2)(O)(OH)]+ with loss of water. Reaction of [Mn(Py2N2)(O)(OH)]+ with a second molecule of H2O2 provides the Mn(IV) oxo/hydroperoxo species [Mn(Py2N2)(O)(OOH)]+. The lowest energy conformation of [Mn(Py2N2)(O)(OOH)]+ is expected to have an intramolecular hydrogen bond between the hydroperoxo and the oxo ligands, which facilitates intramolecular proton transfer, leading to reductive elimination of O2 and formation of the Mn(II) hydroxo/aqua complex [Mn(Py2N2)(OH)(OH2)]+. An acid-base reaction between this species and H2O2 regenerates the aqua/hydroperoxo species [Mn(Py2N2)(H2O)(O2H)]2+ along with H2O.
Scheme 1.
 2. Electrochemical water oxidation
2. Electrochemical water oxidation
In basic solution (pH 12.2), the cyclic voltammogram (CV) of (Py2NtBu2)Mn2+ (Fig. 1, R = tBu) in reveals a catalytic wave that corresponds to an overpotential for water oxidation of ca. 800 mV (Fig. 2). Controlled potential electrolysis experiments with a fluorine-doped tin oxide (FTO) working electrode show catalytic activity for about 100 min. Dioxygen is formed during electrolysis and has been characterized by a number of methods, including an O2 electrode, gas chromatography and the decrease in the solution pH of over the course of the electrolysis experiment. Modest turnover numbers (TON) of 4 – 7 with Faradaic efficiencies of 75 – 84% are observed.
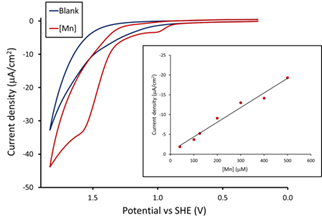
Fig. 2. Cyclic voltammogram for (Py2NtBu2)Mn2+ showing a catalytic current at high potential. Experimental conditions: pH = 12.2, 0.1 M KOTf, FTO working electrode. Inset: Dependence of the catalytic current on catalyst concentration.
Multiple control experiments have been conducted to verify the homogeneous catalytic water oxidation reactivity of this complex. In solution, UV-vis spectroscopy and dynamic light scattering measurements are inconsistent with the formation of nanoparticles. There is also no evidence for the formation of a heterogeneous electrocatalytic deposit on the electrode surface, as determined by CV, optical spectroscopy and energy-dispersive X-ray (EDX) measurements.
The electrocatalytic mechanism has been investigated. Differential pulse voltammetry (DPV) reveals two waves, whose pH dependence has been assigned to the following two reactions:
[(Py2NtBu2)Mn(H2O)2]2+ → [(Py2NtBu2)Mn(H2O)(OH)]2+ + e- + H+
[(Py2NtBu2)Mn(H2O)(OH)]2+ → [(Py2NtBu2)Mn(O)(OH)]+ + e- + 2H+
The catalytic current varies linearly with the concentration of the Mn complex (Fig. 2, inset), consistent with a mononuclear catalyst for water oxidation. Based on the precedent of other single-site electrocatalysts and the first order dependence of the rate on catalyst concentration, we suggest that O-O bond formation involves attack of H2O/OH- (possibly intramolecularly) on the oxo ligand.
This complex is a therefore a bona-fide mononuclear Mn complex for homogeneous electrocatalytic water oxidation in aqueous solution. Many other claims for water oxidation by Mn complexes do not report the appropriate control experiments to test for catalyst homogeneity. The observation of water oxidation by single Mn center provides a functional model for similar single-site catalysis in the oxygen-evolving complex of Photosystem II. 3. Electrochemical water reduction
We previously reported that (Py3NH3)Mn2+ (Fig. 3) shows catalytic activity for electrochemical water oxidation in mildly acidic aqueous conditions. Efforts to investigate the performance of this catalyst are still ongoing. Suspecting that the macrocycle ligand may play a critical role in facilitating water reduction, we have started investigating water reduction catalysis when this ligand is attached to metals that are more widely recognized as catalysts for water reduction. Thus, the analogous Co complex shows very high activity in water reduction, albeit in mixed acetonitrile/water solution (Fig. 4).
Fig. 3. (Py3NH3)Mn2+ in aqueous solution.
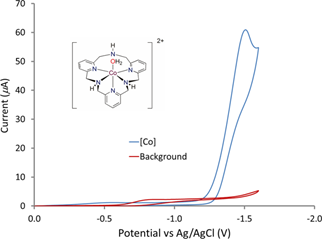
Fig. 4. Cyclic voltammogram for (Py3NH3)Co2+. Experimental conditions: 50% MeCN/H2O, glassy carbon working electrode, 0.1 M KOTf. 4. Career Impact and Student Impact
While at NMSU, PI was a member of a team from three campuses in the state (NM Tech, NMSU and UNM) that aimed to develop methods for the use of solar fuels. The results from the ACS-PRF supported project were used as preliminary data as part of a funded solar fuels sub-project ($2 million) of the New Mexico EPSCoR renewal application. The PI has now moved to a new institution, Indiana University, where his growing expertise in electrocatalysis will be used in joint proposals related to energy research. So far, a paper describing the H2O2 disproportionation results have been published. Our initial paper on water oxidation was rejected and we are currently revising the manuscript for resubmission.
A graduate student is currently employed on the project, beginning August 2012, and will continue to be employed for the upcoming year. This research assistant support will allow him to devote more time to the project.









