Reports: DNI652494-DNI6: Dynamics and Kinetics of Carbon Dioxide in Nano-Porous Materials Environment from Quantum Molecular Dynamics Simulations
 printer friendly
printer friendly
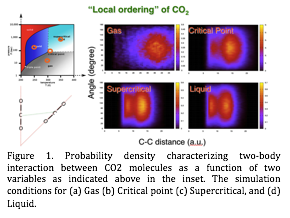
| Figure 1. Probability density characterizing two-body interaction between CO2 molecules as a function of two variables as indicated above in the inset. The simulation conditions for (a) Gas (b) Critical point (c) Supercritical, and (d) Liquid. |
 Significant
portion of the first year of the grant period (9/1/2012-8/31/2013) was spent on
training new graduate students in Density Functional Theory and First
Principles Molecular Dynamics simulations before they could begin the
simulations.
Significant
portion of the first year of the grant period (9/1/2012-8/31/2013) was spent on
training new graduate students in Density Functional Theory and First
Principles Molecular Dynamics simulations before they could begin the
simulations. CO2 molecules in bulk and in confined environments
We employ large-scale first principles molecular dynamics (FPMD) simulations in Car-Parrinello extended Lagrangian formulation as a primary computational tool for investigating detailed dynamics of CO2 in the nanoscale pores. In Car-Parrinello extended Lagrangian approach the potential energy surface is derived "on the fly" from the instantaneous ground state of the electrons. The latter is described within Density Functional Theory (DFT), and PBE exchange-correlation functional is used. We found the planwave expansion of 25 Ryd and 200 Ryd for the wavefunctions and electron density to be sufficient for our investigation here.
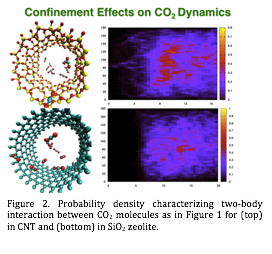
| Figure 2. Probability density characterizing two-body interaction between CO2 molecules as in Figure 1 for (top) in CNT and (bottom) in SiO2 zeolite.
|
 We are also in process of testing dispersion
interaction (van der Waals) in potentially improving the DFT description of CO2
confined in nano-porous environments. We employed the
"van der Walls" correction to DFT in context of the recent atomic pseudopotential approach (Zhang and Duan,
2005), where the van der Walls correction is given in terms of pre-determined
potentials centered on atoms. This computationally efficient approach allows us
to perform long enough FPMD simulations for obtaining statistically meaningful
results. We have adapted this scheme to ultra-soft pseudopotential
in context of FPMD.
We are also in process of testing dispersion
interaction (van der Waals) in potentially improving the DFT description of CO2
confined in nano-porous environments. We employed the
"van der Walls" correction to DFT in context of the recent atomic pseudopotential approach (Zhang and Duan,
2005), where the van der Walls correction is given in terms of pre-determined
potentials centered on atoms. This computationally efficient approach allows us
to perform long enough FPMD simulations for obtaining statistically meaningful
results. We have adapted this scheme to ultra-soft pseudopotential
in context of FPMD. The specific aim at this stage is to characterize how CO2 molecules inside the nano-pores behave in terms of their gas, liquid or supercritical phase behavior by analyzing the diffusivity, pair correlation functions of different atoms, and other dynamical autocorrelation functions. We have already obtained results for bulk CO2 case in different phases, which will serve as a reference for characterizing CO2 behavior in the confined environments. FPMD simulations were run for 30 pico-second after the pre-equilibration time of 3 pico-seconds. Figure 1 shows the probability distribution between two CO2 molecules in terms of the angle and the distance between the carbon atoms as
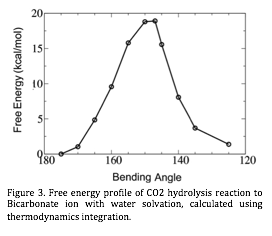
| Figure 3. Free energy profile of CO2 hydrolysis reaction to Bicarbonate ion with water solvation, calculated using thermodynamics integration.
|
 shown schematically. In the gas phase,
there is a single probability distribution centered at 90°/35 a.u.
However, the distribution function starts to show well-defined bimodal
distribution for liquid and super-critical phases at 90°/7 a.u
and 90°/13 a.u.. We are currently investigating how different phases can
be distinguished at molecular level by analyzing various autocorrelation
functions.
shown schematically. In the gas phase,
there is a single probability distribution centered at 90°/35 a.u.
However, the distribution function starts to show well-defined bimodal
distribution for liquid and super-critical phases at 90°/7 a.u
and 90°/13 a.u.. We are currently investigating how different phases can
be distinguished at molecular level by analyzing various autocorrelation
functions.We have recently started investigating CO2 dynamics in carbon nanotube (CNT) and SiO2 zeolite pores of about 1 nm as shown in Figure 2. The comparison of CO2 dynamics in the nanoscale pores of CNT and of the SiO2 zeolite will provide an interesting insight into how the molecular dynamics is influenced by the atomistic details of the inner pore surfaces. The CNT surface is single-atom thick of all sp2 carbon atoms, making its electronic structure rather "homogeneous". On the other hand, the SiO2 inner pore surface exhibits a strong variation in the electrostatic potential, which influences the CO2 dynamics through its polarizability. Although we do not have enough statistics at this point to present any meaningful analysis, the probability density distribution (as a function of the angle and C-C distance) in Figure 2 shows that CO2 molecules approach much closer in the SiO2 zeolite than in CNT. In comparison to all bulk CO2 phases, such a close C-C distance is not observed. As we obtain more statistics, we plan to investigate how the surface polarizability in SiO2 zeolite potential facilitates such CO2-CO2 interactions.
We have also started investigating how water solvation influences the CO2 hydrolysis to bicarbonate ion in bulk water. Using thermodynamics integration approach with FPMD, the free energy barrier of approximately 20 kcal/mol was obtained for the reaction as the OCO angle as the reaction coordinate (Figure 3). Having demonstrated that we can obtain the excellent agreement with the experimental value, we are currently investigating how this barrier height changes in the porous environments. In a confined environment, the solvating water molecules cannot form perfect hydrogen bond network, and it is expected to influence the reaction barrier through the entropic contribution. Understanding how the equilibrium constant for CO2 hydrolysis changes in such a unique environment of nano-scale confinement and high temperature will be a major step in the field and also important for CO2 sequestration.
Reference
Zhang Z.; Duan Z. An optimized molecular potential for carbon dioxide, Journal of Chemical Physics 2005, 122, 214507-214522









