Reports: ND152184-ND1: Organocatalytic Routes to Stereogenic, alpha, alpha-Disubstituted Cycloalkanones
 printer friendly
printer friendlyThe overall goal of this grant is to develop new methods to access alpha, alpha-disubstituted cycloalkanones. At the time this grant was submitted, this project was in its complete infancy. We had conducted a few test experiments; however, we had no major breakthroughs (best result in proposal was just 19% ee).
I am pleased to report we have made major progress with developing the first general organocatalytic method to accomplish this transformation (Scheme 1). Key to this work was the discovery that a primary amine / thiourea dual catalysis system can mediate this transformation. After screening several different catalyst scaffold, we identified the catalysts derived from 1,2-diphenyl-1,2-diamino-ethane were the most promising. Variation of the substituent on the thiourea moiety proved intriguing – as certain substrates worked best with "aniline-derived" thiourea catalysts while others worked best with "benzylamine-derived thiourea catalysts. The preliminary account of this work was published in Organic Letters (Org. Lett. 2012, 14, 3178-3181).
Scheme 1. Summary of Bifunctional Catalyzed Michael Additions.

In follow-up work, we have expanded the scope of the substrates effective by this transformation – including to add some increased steric hindrance at R' position. We have unfortunately to date been unable to mediate this transformation using beta-substituted electrophiles. We have screened a wide array of catalysts, substrates and conditions – including intramolecular examples as described in the proposal – however none of these transformation have proven effective to date.
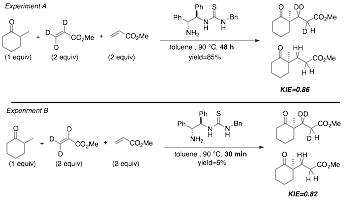
In order to garner a better understanding of this transformation, we have gone on to more thoroughly investigate this nature of this transformation – particularly to explore the reaction mechanism. We had hypothesized that this reaction proceeded through an rate-determining Michael addition step; however, Pfau and dAngelo had indicated in their early experiments that the enamine formation step was possibly rate determining. In order to probe this mechanism, we synthesized a variety of deuterated compounds (Scheme 2). What ultimately proved most effective was the use of deuterated electrophiles as shown below. We were pleased to find a reasonable inverse second kinetic isotope effect (KIE) of 0.82-0.86. Additionally, through our collaboration with Professor Paul Cheong's group at OSU, we have computed the predicted KIE for this transformation, which is in good agreement with our experimental data (0.88 theory vs 0.82-0.86 experimental). Based on this analysis (as well as additional computational work), we feel confident in stating that rate determining step in this mechanism is the Michael addition step.
Scheme 2. Kinetic Isotope Experiments on Bi-functional Catalyzed Michael Additions.
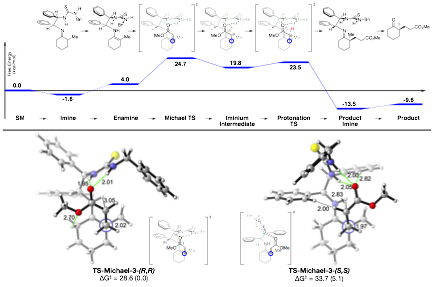
In addition, we have worked with Paul Cheong's group to more fully map out the reaction mechanism for this transformation (Scheme 3). The mechanism proceeds mostly as anticipated; however, computations show an unanticipated resting state of the catalyst on the product imine. Experimentally, we observe the rapid formation of the starting imine structure. Interestingly, we do not observe formation of the enamine by NMR or the product imine. We are unsure as the the exact nature of this divergence; however, we hypothesize that the increased steric requirements of the product imine lead to rapid hydrolysis to as a mechanism to relieve steric compression – likely catalyzed by the neighboring thiourea. This kinetics work as well as the mechanistic analysis is in the process of being written up as a full paper to be submitted to JOC or ACS Catalysis by the end of the year. ACS PRF will be acknowledged for support of this work.
Scheme 3. Mechanism of Asymmetric Michael Reaction.
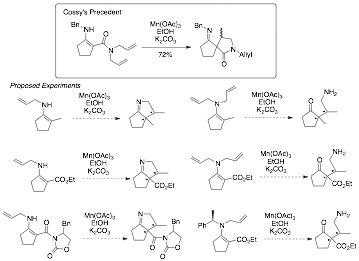
Based on our experimental and computational work, we hypothesize that it is unlikely that an organocatalyzed process utilizing beta-substituted electrophiles (e.g. methyl crotonate) can be developed using enamine-based nucleophiles. That said, we do see an attractive alternative that will be the focus of our future work in the area - utilizing a radical approach to facilitate the key C-C bond. This approach would allow us to build our targeted alpha, alpha-disubstituted cycloalkanones with neighboring stereogenic centers and would circumvent the problematic reactivity we observed in the "two electron" processes we studied. Interestingly, we were surprised to find that radicals of enamines have received only limited investigation to date. Shono and Chiba independently explored electrochemical oxidation of enamines (Shono: BCSJ, 1978, 51, 2179-2180 and Chiba: JOC 1979, 44, 3519-3523). Schoeller and co-workers followed up on this pioneering work 10 years later (JCSPTII, 1988, 369-373). All of these efforts focused exclusively on the electrochemical oxidation of the enamine and not on any subsequent reactivity. More recently, Cossy and co-workers have utilized enamines derived from beta-keto esters in a transition metal and electrochemical-mediated processes to facilitate radical cyclizations – making all-carbon quaternary centers (J. Electroanalyt. Chem. 1996, 389, 215-218; Tetrahedron 1999, 55, 6483-6496; JOC 2000, 65, 7257-7265). This work was limited to a small subset of examples and did not focus on any asymmetric applications. We are just beginning to explore the scope and possibility of this reaction process. Our initial efforts will be directed towards simply exploring the scope of the racemic process; however, we hope to transition to enantioenriched examples – including those derived from alpha-methyl benzylamine. A representative set of initial experiments is shown below. We are really excited about the possibility of opening up completely new reactivity for accessing the target core alpha, alpha-disubstituted cycloalkanones scaffold with neighboring stereochemistry.
Scheme 4. Radical Approach to alpha,alpha-Disubstituted Cycloalkanones.
Synthesis of Tricyclic Annulation Products. Another major area of focus in the proposal was directed towards the development of an asymmetric phase-transfer catalyzed annulation based on pioneering work by Miesch. We screened a range of conditions in using a subset of standard PTC's and bifunctional catalysts (two representative examples shown in Scheme 5) with an array of bases. To our complete frustration, we consistently found that the presence of the PTC inhibited reaction – leading to extensive decomposition. We never could observe formation of the desired tricycle under any asymmetric conditions. A small amount of a spirocyclic compound was observed (typically 7-15%); however, the overall efficiency and catalytic inhibition led us to completely abandon this approach.
Scheme 5. Attempted PTC .










