Reports: ND152167-ND1: Chemical and Conformationally Driven Switches in Covalent Gas Separation Materials
 printer friendly
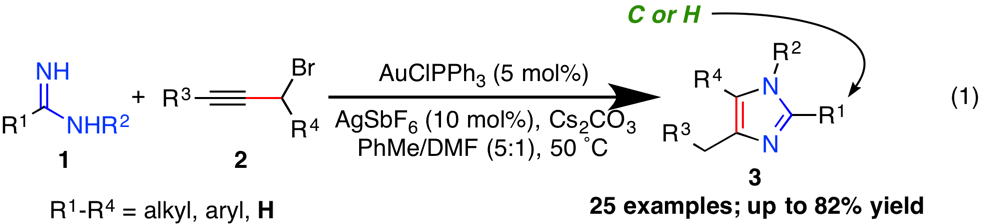
printer friendlyI. Development of a formal Au/Ag-catalyzed [3+2] cycloaddition toward substituted imidazoles.
A major challenge in identifying the optimal material for selective and reversible carbon capture is designing a scalable synthetic approach that allows access to multiple structural motifs with the modular flexibility necessary for point modifications. While our preliminary studies have taken a broad spectrum approach toward heterocycle evaluation to include pyrroles, pyrazoles, and 1,2,3- and 1,2,4-triazoles one focus during this funding was the development of a universal synthetic strategy in imidazole assembly that will allow better flexibility and scalability than conventional approaches. The imidazole ring constitutes one of the most versatile N-heteroaromatics in organic synthesis, and is a well-established precursors to stable N-heterocyclic carbenes (NHCs), which are valuable functional groups for organocatalysts and ligands for transition metal complexes. Especially in the context of materials design and synthesis, imidazoles are exceptionally useful in the construction of ionic liquids in the capacity of the cationic and anionic component. In addition to our findings with imidazolide ionic liquids (ILs), our recent recent work on solid imidazolylidene carbenes prompted us to develop strategy that would enable the rapid construction of architecturally diverse imidazoles. We required a flexible approach toward imidazole construction that would enable the maximum amount of structural diversity at C4 and C5 in a highly convergent fashion.
To that end, we speculated that imidazole assembly utilizing a [3+2] cycloaddition approach from an amidine and alkyne would enable a high degree of structural flexibility with optimal synthetic efficiency, while also tolerating C2-alkyl substitution and formamidines in the cycloaddition. This would provide us with the desired structural flexibility we sought along with the ability to directly assemble imidazoles that are readily amenable to conversion to their corresponding N-heterocyclic carbenes. Our efforts have resulted in a streamlined method involving the alkylation of amidine 1 with propargyl halide 2 followed by Au(I)/Ag(I)-catalyzed 5-endo-dig cyclization and C–C double bond isomerization to directly assemble imidazole 3 (Eq 1). This study constitutes a unique new method in imidazole construction that provides direct access to imidazoles with a diverse structural configuration along the C4/C5 backbone. A manuscript is in preparation and will be submitted in the near future. To date, we have assembled a diverse collection of imidazoles in a highly convergent fashion. Notably, the reaction tolerates formamidines as substrates to directly construct C2-unsubstituted imidazoles, which are highly desirable as N-heterocyclic carbene precursors. Future work on the utilization of this method for the construction of a portfolio of energetic materials based on the imidazole framework and further mechanistic studies are currently underway and will constitute our efforts over the next funding period.
II. Development of a formal Au/Ag-catalyzed [3+2] cycloaddition toward substituted imidazoles.
Our remaining efforts during this funding period were directed toward the design and synthesis of a heterocyclic framework affixed with a receptor functionality that will ultimately enable electro/photochemical gas absorption/desorption control. Although Nature does not offer a model of dealing directly with post-combustion CS, there are numerous examples of chemical triggers used in biosynthetic pathways. The installation of excitable functionality on a heterocyclic scaffold would minimize the energetic penalty for regeneration by the electro- or photochemically controlled capture and release of CO2. Based on recent findings by Yam and co-workers that showa weakening of the metal-carbene carbon bond upon a UV light initiated 6¹ electrocyclization of a bis(thienyl) metal carbene complex, we focused our initial efforts on the synthesis and evaluation of a bis(thienyl)imidazole. By targeting this heterocyclic framework, we can access either the corresponding NHC or IL.
Following a protocol developed by Bielawski and coworkers, bis(thienyl)imidazole was synthesized in two steps from commercially available 2,5-dimethylthiophene, and converted into the corresponding IL 4 by deprotonation and anion exchange with P666(14)Br. Unfortunately, alkylation of the parent imidazole with methyl iodide followed by treatment with based failed to provide the corresponding NHC. In an effort to preliminarily evaluate its efficiency at carbon capture, exposure of IL 4 to 1 atm of CO2 yielded the corresponding the carboxylated adduct in near quantitative yield. Subsequent examination of the imidazolide IL 4 as a photoswitchable carbon capture material through a series of electrocyclization experiments revealed that the parent IL 4 would undergo cyclization when exposed to UV light (λ 250-255 nm) (Figure 1). The extent of carboxylation at different temperatures and pressures of both the open and cyclized forms of IL 4 was measured by obtaining an isotherm profile for each through our collaboration with the Brennecke group at the University of Notre Dame. Although promising, additional studies are needed at this time to provide a complete picture of carbon capture efficiency. Future during the upcoming funding period will focus on this evaluation and a systematic assembly of other materials bearing the dithienyl ethene subunit.

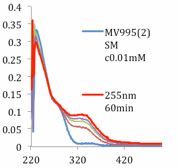
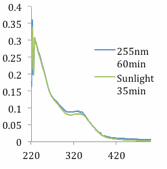
Figure 1: UV/Vis spectra of photoinduced cyclization and reverse reaction of IL 4.
In summary, we have shown that the bis(thienyl)imidazole system is a suitable model system to establish a proof of concept toward the general applicability of our photoswitchable CCS concept. Work that is currently underway for the present funding period include a critical evaluation of the isotherm data at different pressures of CO2 and expanded calculations to include alternate heterocyclic designs. This concept of electro/photochemically-mediated carbon capture will drive future CS research toward the development of new materials that require a minimal parasitic energy penalty for regeneration. These results have had a dramatic impact on defining a new area of research in my group and has facilitated renewed opportunities for the students involved in the project.









