Reports: UNI350877-UNI3: Iron(III) Oxidation of Carboxylic Acids and Phenols as Models for the Abiotic Transformation of Natural Organic Matter
 printer friendly
printer friendlyThis proposal investigates the interaction of iron(III) with natural organic matter (NOM) by using model systems to explore reactions that modify NOM structure. Two aspects of this project have been actively supported by the PRF in the past year and will be discussed separately in this report. These sub-projects are 1) the completion of the study on the mechanism of hydroquinone oxidation by iron(III)-m-oxo dimers supported by tris(pyridyl)methylamine (TPA) ligands, and 2) the initial development of an iron(III)/picolinic acid system that catalytically degrades phenol using O2 and light. One undergraduate student was supported by funds from the PRF last summer and the PI presented the work at a local meeting of the Susquehanna Valley section of the ACS in May.
Impact
Phenols are an important functional group in NOM that undergo oxidative degradation during the humification process. Spectroscopic evidence suggests these moieties are often associated with iron(III) in natural waters. This work broadly investigates the oxidative transformation of phenol and related derivatives by small-molecule NOM analogs that function as stabilizing ligands for diiron(III) m-oxo complexes. Tris(2-pyridylmethyl)amine (TPA) is especially relevant because of the rich history of this ligand supporting diiron(III) complexes that model the active sites of metalloenzymes (e.g. sMMO and RR). Because the aqueous coordination chemistry of diiron(III) TPA complexes has not been reported, this work also contributes to the coordination chemistry of an important bioinorganic model system.
Progress
Iron(III)-TPA complexes
During this reporting period, a substantial amount of time has been spent improving the speciation model in the iron(III)-TPA system. This was done by non-linear least squares fitting of potentiometric titration data and the preliminary results from 2012 has been refined for publication. This process generates formation constants for each species present in the model, which can then be used to quantify the iron complexes present as a function solution conditions (pH, concentration of reagents). Figure 1 shows the composition of the iron(III)-TPA system under the conditions of interest and is consistent with crystallographic data in that the iron(III)-TPA complexes exist as μ-oxo dimers with the general formula [(TPA)FeX]2O (1a: X = H2O; 1b: X = H2O, OH-; 1c: X = OH-)

Figure 1. pH dependent speciation of iron(III)-TPA complexes.
Conditions: 0.99 mM Fe+3; 10.0 mM TPA, 25°C, I = 0.100 M.
The information gained on the thermodynamics of iron(III)-TPA complexes was then applied to the oxidation of hydroquinone, and during the current reporting period we have considerably expanded the kinetic dataset to include lower pH and varying hydroquinone concentrations. We have observed Michaelis-Menten-type pre-equilibrium behavior at pH 5.6 with Kd = 50 ± 4 mM and k2 = 0.61 ± 0.03 s-1 however recent kinetic experiments have revealed a change in mechanism at lower pH. Figure 2 shows a deviation from pseudo 1st-order kinetics at pH 4.63. Similar results were obtained for all experiments below pH 5.6.
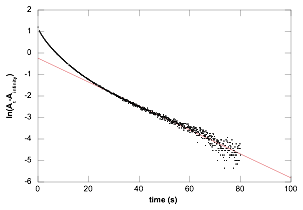
Figure 2. Log plot of integrated absorbance from 485-505 nm. Δt = 9 ms;
[Fe+3]o = 0.99 mM; [TPA] = 10.0 mM; [HQ]o = 59.9 mM. pH = 4.63
These data are suggestive of a system in which two unique species convert to the same product at different rates. We have hypothesized that an acid-catalyzed isomerization of the iron(III) μ-oxo dimer is occurring to relieve the steric congestion caused be replacement of a coordinated water molecule with hydroquinone (Figure 3). This is especially interesting given the variety of TPA coordination modes in iron(III) μ-oxo dimers that have been observed crystallographically. Substitution of hydroquinone for phenol in the system shuts down reduction of iron(III) and produces species that appears to be an iron(III) phenoxide by UV-vis spectroscopy. We are attempting to grow single crystals of this species for x-ray diffraction to support our mechanistic hypothesis.
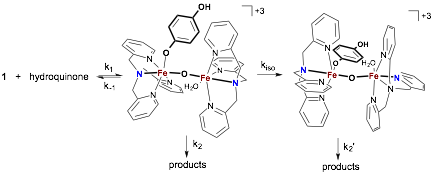
Figure 3.
Proposed mechanism for hydroquinone oxidation by 1.
Isomerization (kiso) is kinetically competitive
at pH < 5.6.
Aerobic Phenol Oxidation
During the current reporting period we have also developed a catalytic system capable of phenol oxidation by O2 using iron(III), picolinic acid (pyridine-2-carboxylic acid) and ultraviolet light. This is an important result given the desirability of using molecular oxygen in industrial oxidations, and also because O2 is the dominant oxidant of NOM found in surface waters. Initial experiments have shown loss of 62% phenol with concomitant formation of 5% hydroquinone and 8% catechol as measured by HPLC. The poor mass balance in this reaction is due to secondary oxidation of hydroquinone/catechol to lead ultimately to ring-opened carboxylic acid products and/or CO2. This was confirmed by calculating the total O2 uptake my monitoring the change in chemical oxygen demand (COD) of the system during photolysis. Note that 7 equivalents of O2 are necessary to affect complete mineralization of phenol to carbon dioxide. No trace of phenol oxidation is observed in the absence of iron, piconlinc acid, or UV light. Continuing work focuses on optimization of the reaction conditions and probing the structure of the iron(III)-picolinic acid complexes responsible for oxidation.
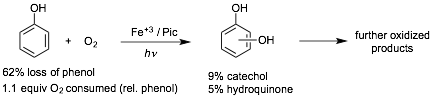
Figure 4. Aerobic
photo-oxidation of phenol catalyzed by iron(III) picolinate.
Initial conditions: 5.00 mM phenol; 0.20 mM picolinic acid; 0.050 mM Fe(NO3)3.
O2 sat. solutions irradiated at 350 nm for 24 hours.
Student impact
This PRF-UNI grant has supported one undergraduate student, Jessica Britton '16, during the current reporting period by providing the funds necessary to allow her to work on the iron/picolinic acid project during the summer of 2013.









