Reports: ND151929-ND1: Self-Assembly of Catalysts and Ligands by Reversible Covalent Interactions of Organoboron Compounds
 printer friendly
printer friendlyLibraries of chiral phosphines prepared by three-component condensations of 2-formylphenylboronic acid. Self-assembly of chiral phosphine ligands through boronic acid–diol interactions was among the initial aims of the project. Among the hurdles encountered was the non-trivial synthesis of boronic acid-functionalized phosphine components. Given the eventual goal of preparing libraries of ligands, straightforward access to the partners for the self-assembly was viewed as a priority. We thus turned our attention to a modified strategy based on three-component condensations of amines, diols and 2-formylphenylboronic acid to generate imine–boronates. These three-component couplings are rapid and high-yielding, and have been used for high-throughput enantiopurity determinations by NMR. Of particular importance for our envisioned application in catalyst self-assembly was the ready availability of chiral, enantioenriched amine-functionalized phosphines. The three-component nature of the couplings was appealing from the perspective of combinatorial library generation, and the envisioned tetracoordinate imine–boronates were expected to be relatively unreactive towards transmetallation, a pathway that could lead to ligand decomposition.
Scheme 1. Self-assembly of a bis(phosphine) ligand by imine-boronatecondensation.
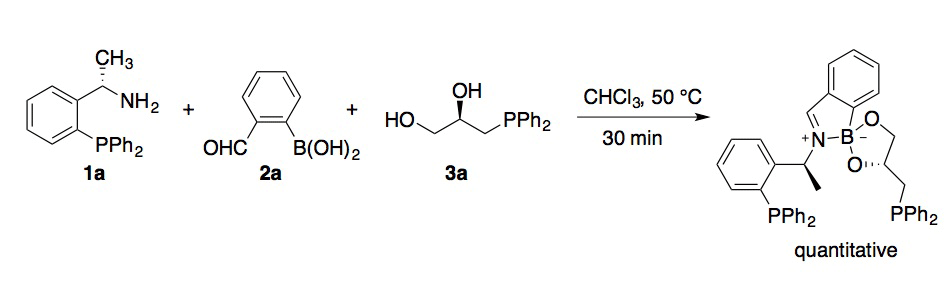
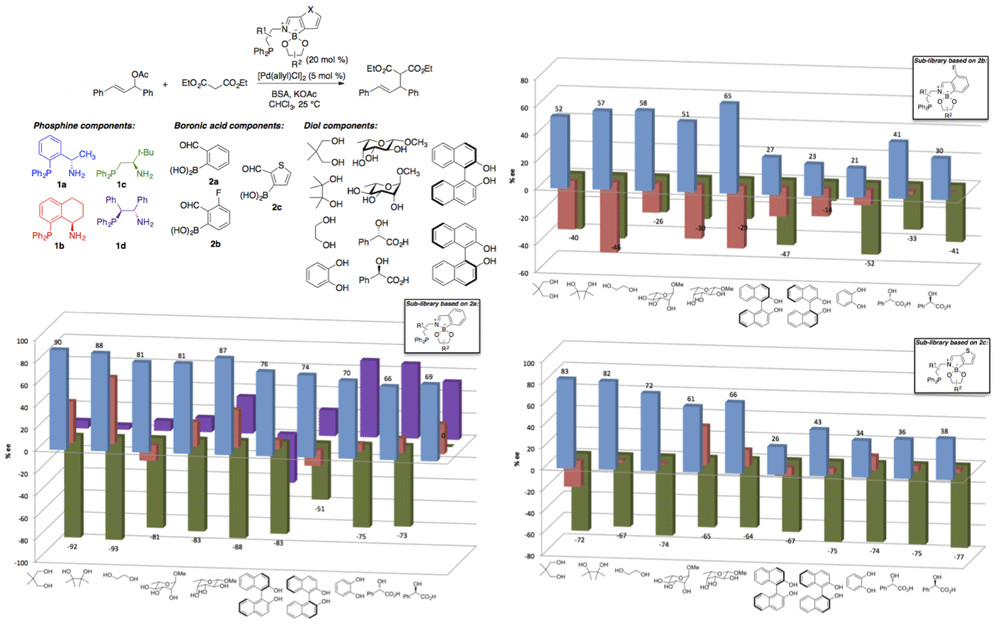
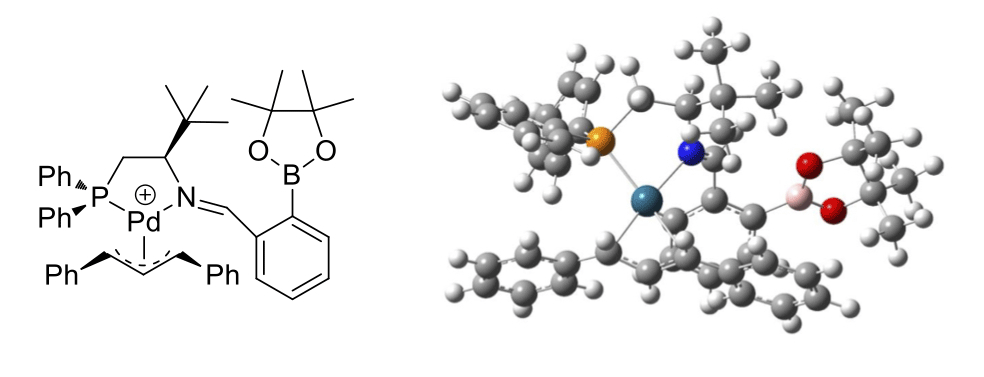
Condensation of phosphine-bearing amine 1a and diol 3a with 2-formylphenylboronic acid (2a) was accomplished in quantitative yield from a 1:1:1 molar ratio of the components in chloroform at 50 ¡C (Scheme 1). Preliminary studies of the Pt(II) complex of the resulting ligand indicated that only one phosphine group was involved in metal complexation, and suggested a P,N- or monodentate binding mode involving the amine-phosphine-derived donor. Ligands assembled by three-component condensation were tested in Pd(0)-catalyzed allylic alkylation reactions. Evaluation of a small set of ligands in which the phosphine moiety was deleted from either the amine or the diol component (data not shown) further pointed towards a P,N-binding mode. On this basis, a library of 100 phosphines was generated from aminophosphines 1a–1d, formylarylboronic acids 2a–2c and diols 3b–3k, with the latter components not possessing phosphine groups. The results of the asymmetric allylic alkylation using this ligand library are depicted in Scheme 2. The enantiomeric excess was sensitive to the structures of all three components, with matching/mismatching effects being apparent for (R,R)- vs (S,S)-BINOL and for amine-phosphine / formylarylboronic acid pairs (1b/2a vs 1a/2b). The ligand generated from tert-leucine-derived phosphine 1c, 2a, and pinacol (3b) provided a 93% ee in the test reaction. Computational modeling of a putative Pd(II)-allyl complex derived from this ligand (Scheme 3) was consistent with P,N-complexation, and suggested that the B–N interaction that helps to drive the 3-component condensation may not persist upon metal coordination. As our group has not previously carried out this type of combinatorial effort, the construction and evaluation of this initial library has provided invaluable experience and expertise that we will exploit in our ongoing efforts.
Scheme 2. Evaluation of a library of 100 self-assembled P,N-ligands for asymmetric allylicalkylation.
Scheme 3. Calculated structure of the Pd-allyl complex derived from a representative self-assembled ligand. Calculations were carried out with Gaussian 09, using DFT with the B3LYP functional and a mixed basis set (LANL2DZ for Pd, 6-31G(d) for N, P and the diphenylallylfragment, and MIDI! for all remaining atoms).
Current work is aimed at varying the electronic properties of the phosphine group, employing this class of P,N-ligands in more challenging allylic substitution reactions, and expanding their applications to other types of transition metal-catalyzed reactions. Although certain limitations have become evident – for example, attempted allylic substitutions using amine nucleophiles resulted in exchange with the imine–boronate – the self-assembled ligands appear to be stable to diverse transition metals and reagents. While efforts towards the initially stated goal of targeting bis(phosphine) ligands continue, exploring less well-developed classes of donors (e.g., sulfoxides, alkenes) will be a focus of our future investigations in this area. The graduate student who has carried out this work presented his results at the 2013 OMCOS meeting, and we anticipate submitting an initial manuscript by late 2013.
Bifunctional catalysts generated by imine–boronate self-assembly. We have used the strategy for modular assembly discussed above to generate hydrogen bond donor-functionalized phosphines. Bifunctional species of this type have been used as Lewis base–Bronsted acid organocatalysts for the Baylis–Hillman reaction and its variants. The ability to systematically tune the properties of the two functional groups, and to expand upon the relatively limited structural range of such bifunctional catalysts that have been employed to date, were viewed as potential advantages of the self-assembly approach. Urea- and thiourea-bearing diols were readily accessed from serinol and epichlorohydrin, and participated in the three-component couplings to generate the target bifunctionalspecies (Scheme 4).
Scheme 4. Representative bifunctional organocatalysts prepared by imine–boronateself-assembly.
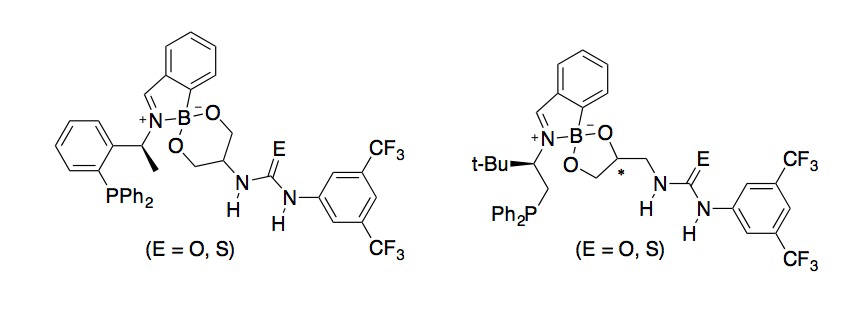
Preliminary evaluation in aza- Baylis-Hillman reactions of N-sulfonylimines with methyl vinyl ketone (data not shown) indicated a beneficial effect of the (thio)urea group on yield in certain instances, but enantioselectivity was low. Evaluation of diverse architectures to link the phosphine and hydrogen bond donor groups is likely needed. Ligands of the type shown in Scheme 4 will also be evaluated in transition metal-catalyzed reactions, as the incorporation of a hydrogen bond donor group into the 'outer sphere' of a metal complex may give rise to interesting reactivity or selectivity.









