Reports: DNI651052-DNI6: Theory of Heterogeneous Oxidation of Carbon Monoxide by Sub-nano Surface-Deposits from Electronic Structure to Catalysis
 printer friendly
printer friendlySmall clusters of transition metals deposited on supporting surfaces are among the most promising but least understood catalytic materials. Their catalytic properties depend unpredictably and nonlinearly on cluster size and composition, and cannot be extrapolated from those of the extended surfaces or lager nanoparticles. We use and develop a full array of multi-scale modeling techniques, ranging from electronic structure to statistical mechanics, to gain insight into these dependencies, and enable rational design of highly active surface-deposited clusters.
Here are our accomplishments resulted from the 2 years of PRF support.
New methodology:
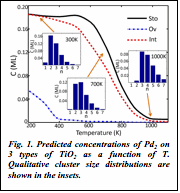
1) We developed a kinetic Monte Carlo method for simulating the process of sintering of deposited clusters via the mechanism of Ostwald ripening. We are now able to predict cluster size- and composition-distribution over a range of temperatures (phase diagrams) in agreement with experimental data (Fig. 1). Using this method we can predict which clusters are actually relevant to potential industrial catalytic processes, and bridge model studies of such catalysts with realistic catalysis.
| Fig. 1. Predicted concentrations of Pd2 on 3 types of TiO2 as a function of T. Qualitative cluster size distributions are shown in the insets.
|
2) We developed a statistical mechanical modeling of catalytic solid/gas interfaces at high temperatures and pressures. This is a QM/MM Monte Carlo method facilitated by the particle filter algorithm from robotics. It is extremely parallel, operating on Argonne supercomputer, utilizing as many as 20,000 cores at once. The method permits for the treatment of heterogeneous interfaces explicitly, at high temperatures and pressures. It is unprecedented in computational catalysis. The manuscript is currently in preparation.
| Fig. 2. QM/MM MC method. The QM-MM boundary goes between the first and second solvation shell, and inside the support material. The rest of the support is seen as a sea of charges, and the solvent is classical. |
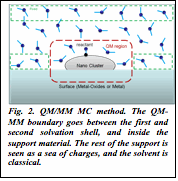
3) QM/MM MC is also our newest and most efficient method for finding the global minima on the potential energy surfaces of clusters, both in the gas phase and on supporting surfaces.
Applications:
4) We assessed structure, stability, and mobility of selected small Pd clusters on titania surfaces. We discovered that these cluster preferentially bind to stoichiometric surfaces, and they are rather mobile on it. O-vacancies serve as migration stoppers, and also facilitate cluster dissociation.
3) We realize electronic reasons for preferred structures of deposited clusters, and develop a predictive qualitative theory of chemical bonding for surface-deposited clusters, to have an insight into their shapes, stabilities, and properties. In particular, we found that clusters change shape upon deposition, from 3D in the gas phase to 2D. Matching with surface O atoms for partially covalent bonding, and additional acquisition of the aromatic character of chemical bonding drives the transition. This is the first time aromaticity was discovered in surface-deposited clusters.
4) We study the activity of these clusters toward the reaction of CO oxidation as a function of cluster size. We now recognize that the key to the catalysis in the parallel experiments by Prof. Anderson (U of U) is the activation of oxygen, which is done better by clusters that permit multi-dentate binding of oxygen. Additionally, the process can be facilitate by the nearby O-vacancies that dissociate O2.
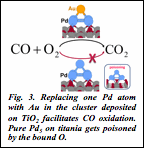
5) Our insight into the electronic structure of these systems allowed us to rationally choose Au as a low-concentration dopant for deposited Pd clusters, for more efficient catalysis. The smallest such cluster, AuPd4 on titania is more efficient at CO oxidation than Pd5 on titatnia. The effect is due to smaller affinity to O2, which therefore cannot poison the catalyst, as in the case of pure Pd clusters (Fig. 3).
| Fig. 3. Replacing one Pd atom with Au in the cluster deposited on TiO2 facilitates CO oxidation. Pure Pd5 on titania gets poisoned by the bound O. |
6) In parallel with this effort, we also pay close attention to gas phase clusters and new aspects of chemical bonding in them. For example, we discovered that hybridization of atomic orbitals can be present in all-metal clusters and impact their shapes. Furthermore, the effect of hybridization on shape is opposite of that of delocalized (aromatic) bonding. Therefore, the two effects constitute the "knobs" of cluster design. This work is primarily done with undergraduate researchers.
| Fig. 4. Aromatic and ionically bound clusters contain tetracoordinated square planar C or Si in the global minimum structures. Covalent structures are instead distorted, C2v. |
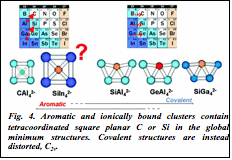
7) We used this discovered mechanism to design a new cluster containing a tetracoordinated square planar Si atom, for example (Fig. 4). As a project of opportunity, along these lines, we discovered a new molecular motor, the B13+ cluster. It possesses a dual ring structure, and the inner ring can rotate with respect to the outer ring when the cluster is exposed to the circularly polarized light in the THz range.
8) The current applied focus is on larger clusters of Pd and Pd doped with Au and Cu deposited on titania for CO oxidation; and doped Pt clusters deposited on titania, magnesia, and silica as catalysts for selective dehydrogenation.
The grant was used to support my postdoc, Jin Zhang, and partly my graduate student, Elon Weintraub, and partly my summer salary. Grant was also used for travel to conferences: ACS Meeting, and Gordon Conference on Catalysis.
This PDF ND grant helped me establish a new direction in my lab dedicated to computational studies of surface-deposited clusters in catalysis, and realistic multi-scale modeling of such heterogeneous interfaces. I now won a larger AFOSR grant to continue the effort and use the new methodology developed under the support of ACS PRF.









