Reports: DNI152447-DNI1: Metal Catalyzed C-H Bond Activation and Allyl Addition: Development of New Transition Metal Catalysis for Practical and Uniquely Efficient Carbon-Carbon Bond Forming Reactions
 printer friendly
printer friendlyThe focus of this funded proposal is the development of new metal-catalyzed, allyl addition reactions between simple alkene hydrocarbons (nucleophile) and carbonyl groups (electrophile). The research conducted with financial support from the PRF aimed to link two distinct reactions: (i) metal-catalyzed allylic C–H activation, and (ii) carbonyl allyl addition. The net transformation corresponds to a carbonyl allylation with control over site-selectivity but without the need to first synthesize the metal-allyl reagent. Such transformations offer a highly attractive and an entirely different strategic alternative to the approaches involving pre-formed organometallic reagents. Importantly, this merger couples C–H bond cleavage with C–C bond formation in addition to generating up to two new contiguous stereogenic centers with a proximal alkene; the alkene unit provides a handle for further transformations. If successful, these new catalytic processes will provide 100% atom-economical approaches to a number of secondary and tertiary homoallylic alcohols that will be of significant utility.
Our proposed studies were based on palladium catalyzed allylic C–H activation to generate metal p-allyl complexes (i ¨ ii) from the corresponding alkenes (complexes that can exist as either h1- or h3-), which then add as a nucleophile to a variety of C=O electrophiles.
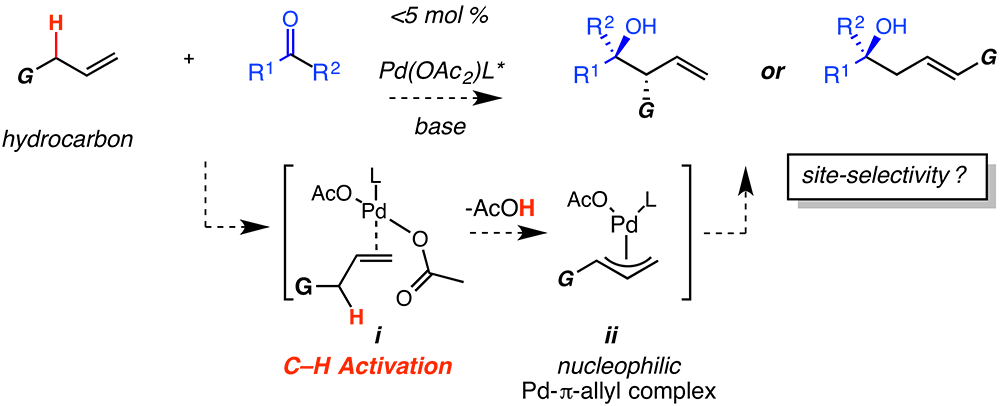
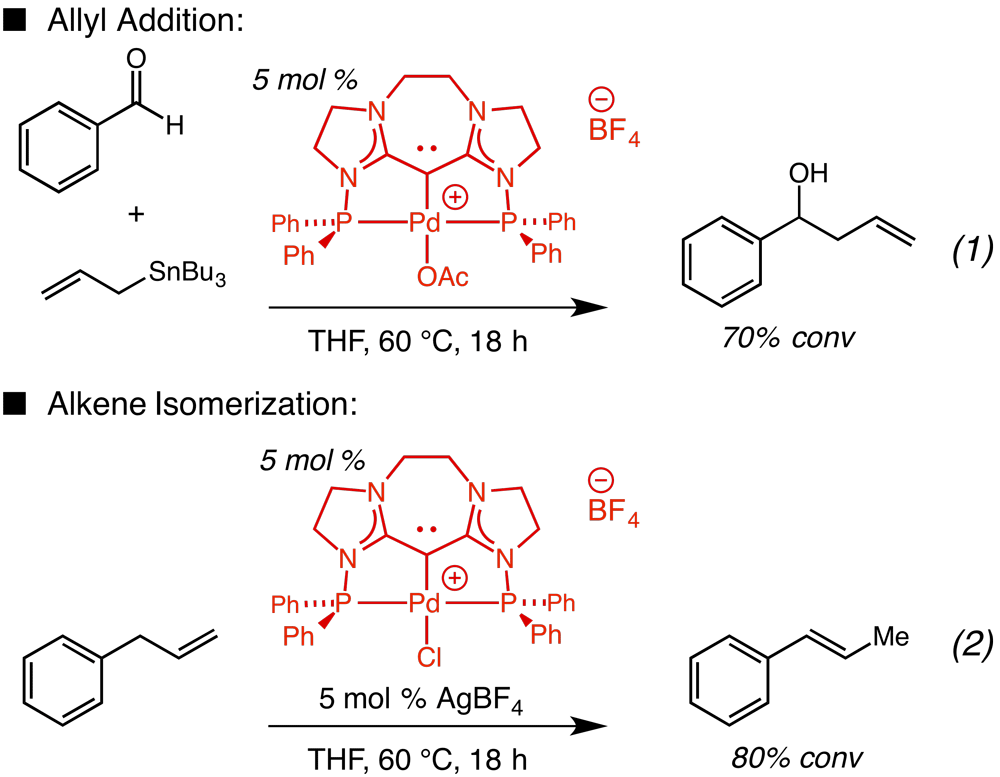
Our initial approach was based on the ability of palladium catalysts to facilitate allylic C–H bond activation as well as catalyze addition of allyl groups to carbonyls. However, exploitation of transition metal-catalyzed C–H bond functionalization in this context requires the action of a catalyst with the correct chemical attributes that serves to: (1) break the C-H bond, and (2) control the reactive nature of the resulting organometallic species. One of the most significant challenges in developing this process is the need to identify a ligand or small sub set of ligands for palladium capable of promoting both electronically distinct key steps in the desired catalytic process. We first investigated N-heterocyclic carbene (NHC) Pd complexes due to their known ability to promote catalytic allylation. Initial studies revealed NHC-palladium complexes to be ineffective for C–H/allylation, in part due to benzoin side reactions observed due to free NHC in solution. After an unsuccessful ligand screen we decided to move towards developing alternative electron-donating ligands, for example carbodicarbenes (carbon(0) ligands); such ligands exhibit unique properties, which can also be sterically and electronically adjusted by easy modification of the ligand scaffold. We have successfully synthesized and developed a class of tridentate carbodicarbenes that form well-defined palladium complexes that can be easily modified with a single reactive coordination site. We reasoned that a tridentate scaffold would promote formation of an h1-allyl fragment capable of allyl transfer. Preliminary studies indicate that the corresponding cationic palladium(II) complexes bearing the new tridentate carbon(0) ligands are effective catalysts for (i) aldehyde allylation with allyl-metal reagents and (ii) alkene isomerization. An allyl metal reagent was used to test the C–C bond forming capability of the Pd catalyst. As illustrated, 5 mol % of a Pd-carbodicarbene complex promotes allyl addition with an allyl-stannane (Eq. 1). In order to model the C–H activation step we first looked at catalytic alkene isomerization; such a process that should proceed through a Pd-allyl complex. We found that the cationic Pd(II)-complexes facilitate rapid isomerization of alkenes at 60 °C, as represented by the isomerization of allylbenzene to b-methylstyrene (80% conv, Eq. 2). Though we have been able to demonstrate that the new carbon(0) Pd complexes are capable of promoting both parts of a C–H activation/allylation catalytic cycle we have yet to demonstrate coupling of both sections. We are currently trying to identify whether the isomerization proceeds through a Pd-h1-allyl fragment as well as continuing to progress towards a unified catalytic C–H activation/allylation process.
Our studies have highlighted the challenge in developing a catalyst with a metal center that is required to carry out two electronically distinct transformations. In addition to developing new carbon-based ligand scaffolds, we are pursuing an additional strategy for C–H/allyl additions. Interestingly, this approach involves developing a bi-functional-type catalyst that does not require the metal center to promote both transformations, C–H bond cleavage and nucleophile addition, required in a C–H/allylation manifold. Developing bi-functional metal catalysts is an approach we are currently exploring in addition to developing the tridentate carbodicarbene ligands.
The funding provided by the PRF has allowed us to develop our C–H/allylation program that we believe will provide a catalytic strategy for the direct conversion of alkenes into allyl nucleophiles; these reagents important for the synthesis of homoallylic alcohols. Our work has led to the design of a new class of tridenate carbon(0)-based ligands for transition metal catalysis. Although, we have not yet realized a catalytic C–H/allylation reaction we are continuing to make progress to solve this problem. Furthermore, the properties of the carbodicarbene ligands, scaffolds, and metal complexes developed in this program have provided us with exciting new avenues of research to pursue. We plan to publish the results of our studies on the new carbodicarbene-based ligands and reactions promoted by their corresponding metal complexes. My graduate student, who has been the main contributor to our efforts, recently competed successfully for an NSF graduate fellowship as well as passed her second year preliminary oral examination based on her research supported in part by this grant. Our continuing efforts in advancing a C–H activation/allylation program remains one of the foundations of our group's research.









