Reports: UNI151776-UNI1: Development of Oxyallyl Silanes as Homoenolate Equivalents in the Oxidative Addition to Vinylogous Amides
 printer friendly
printer friendlyOverview:
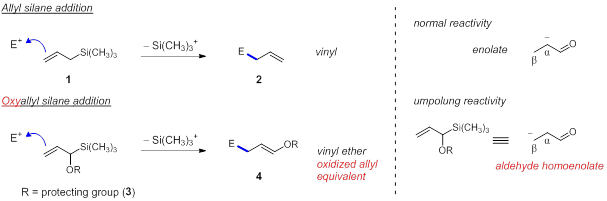
Currently our research group is pursuing the development of methodology using oxyallyl silanes as aldehyde homoenolate equivalents in the addition to various electrophiles. A standard addition of allyl silane to electrophiles generates a product containing a three carbon chain and a terminal vinyl group (1→2). Oxyallyl silane addition furnishes a three carbon chain plus the terminal vinyl group at an increased oxidation state as a vinyl ether, which serves as a masked aldehyde (3→4). This precludes the use of traditional methods for post-functionalization of an alkene such as hydroboration-oxidation which can be difficult when the compound contains a nitrogen or when the alkene is near a sterically hindered position. The vinyl ether is already at the desired oxidation state to allow for further conversions or an intramolecular cyclization reaction using only a protic acid. The initial results of this work will be described here including the progress made towards synthesizing protected oxyallyl silanes derivatives (3) and developing a new route to synthesize oxyallyl silanes starting from readily available precursors.
Summary of Experimental Work:
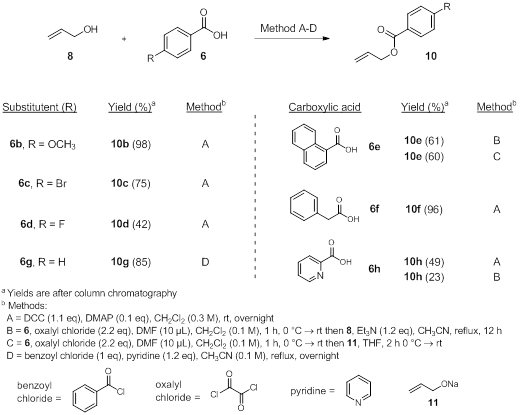
The synthesis of oxygen-protected oxyallyl silanes is sought to prevent undesired side reactions with the free alcohol and the electrophiles to be studied. Aromatic esters were chosen for their ease of synthesis and ability to identify readily using UV light. The step to synthesize esters 3 began with hydroxy allyl silane 5. A DCC coupling reaction with aromatic carboxylic acids (6a-f) using catalytic DMAP provided esters 7a-f in varying yields. After the reactions were complete, the urea byproduct from DCC was filtered and the crude reaction was chromatographed on silica gel. One problem is that the urea does not always filter out completely and makes the reaction difficult to purify. A second difficulty is a side product formed from incomplete esterification involving an adduct of 6 with the DCC. This product co-elutes with some of the esters requiring two columns to obtain a high purity of 7.

In addition, our supply of hydroxy allyl silane 5 was running low. However, the most common route to synthesize 5 requires the use of the extremely flammable and unsafe tert-butyllithium (tert-BuLi). The one pot reaction begins with allyl alcohol 8 which is deprotonated using n-BuLi and then reacted with TMSCl to form 9. In the same pot, 9 is exposed to tert-BuLi which deprotonates one of the allylic hydrogens and causes the retro-[1,2]-Brook rearrangement to take place in which the silyl group moves from the oxygen to the carbon. Upon workup, 5 is formed in moderate yields. Our group is currently trying to find a robust method to synthesize 5 on large scale using safer reagents, especially in an undergraduate setting.

In the meantime, we also optimized the coupling reaction to form aromatic esters using allyl alcohol as a model system (8). Four different methods were attempted (8→10, as in the table below). The DCC method (Method A) provided similar results and difficulties as mentioned before, even without the silicon group present. To try and optimize 6e and 6h, two additional methods were attempted. These included making the acid chloride of 6, which avoided the use of DCC. Method B used allyl alcohol and Method C used the sodium alkoxide of allyl alcohol (11, made by reacting 8 with NaH in THF). Neither method showed an improvement compared to DCC but a more thorough screen of these conditions with more substrates is currently ongoing.
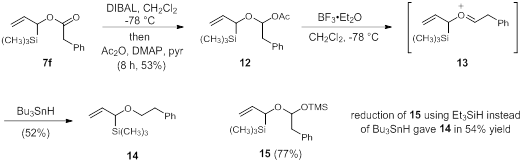
Other protecting group derivatives of oxyallyl silanes (3) are also desired in addition to esters since the goal is to screen how 3 will add to electrophiles. Esters may prove to have an electron withdrawing effect on the alkene and slow the addition of 3. As such, we also pursued ether derivatives. Using the Rychnovsky protocol, the esters can be converted to ethers in two sequential reduction steps – reduction to an acetate protected acetal (12) and then reduction to the ether (14). This conversion was attempted on ester 7f. DIBAL reduction and quenching at cold temperatures with acetic anhydride provide acetal 12. Exposure of 12 to a Lewis acid (BF3•OEt2) forms oxonium ion 13 and then adding a hydride source (tin hydride) gave the ether in moderate yield. The reduction method is inconvenient to run in an undergraduate setting due to the long times required for the first reduction step. Since the acetate is lost in the end, we pursued alternative quenching reagents. We found that TMS-imidazole could be used instead of acetic anhydride resulting in reduced time (8 h to 2 h) and an increased yield (15, 77%). In addition, Et3SiH could be substituted for Bu3SnH (which is toxic and difficult to remove during purification) and the yield was still maintained. This reaction, however, did not work for aromatic esters (7a-7e) and only proved successful for non-aromatic ester 7f (contains a phenyl ring that is not attached to the ester). The development of a method for the conversion of aromatic esters to ethers is currently underway in our group.
Undergraduate Student Impact:
This grant allowed me to support four community college undergraduate students on summer stipends for research. These same four students gave a total of eight presentations at local conferences (Queensborough Honors Conference and the NY ACS Undergraduate Research Symposium) and a national venue (244th National ACS Meeting in Philadelphia, PA). The students supported were diverse (Hispanic, Asian, black, and female) and all plan to continue their education in the STEM or healthcare fields. Two plan to continue on in chemistry, one in chemical engineering, and one as a Physician's Assistant. The PI and her students at Queensborough Community College benefited greatly from funding from the Petroleum Research Fund. During the award period, the PI moved to a new institution (University of Southern Mississippi) which was not supported by the UNI undergraduate funding category. She will continue her research at USM with a focus on collaborations between local community colleges.









